Martell Winters, Director of Scientific Competency, and Wendy Wangsgard, Ph.D., Senior Scientist—Nelson Laboratories07.23.18
A new version of the ISO document regarding bioburden testing was recently published. Its designation is ANSI/AAMI/ISO 11737-1:2018—Sterilization of health care products—Microbiological Methods—Part 1: Determination of a population of microorganisms on products. Though the document has a number of changes, this article will highlight the top five things manufacturers need to know.
#1: Bioburden Method Suitability
Bioburden test methods are dependent on the ability of microorganisms to replicate in the bioburden test system. Some products tested for bioburden can release substances that inhibit microorganism replication. The inhibitory substance is typically known to the manufacturer because it is intentionally included as part of the product (i.e., chemical or antibiotic). Sometimes, however, an unknown inhibitory substance can be present that can be problematic because the manufacturer may believe there is no need to test for inhibitory substances. For example, there might be residuals from cleaning or disinfecting processes on the product, or on components of the product that are provided by a supplier.
When an inhibitory substance is present, the bioburden test results can look very low (e.g., 0 CFU), even though there are viable microorganisms on the product. This is because inhibitory substances sometimes do not inactivate or kill the microorganisms; they merely inhibit them from replicating.
There is an analogous test used in qualifying a test of sterility. It has historically been called the Bacteriostasis/Fungistasis test or B/F test; it is currently called Method Suitability. The intent behind that test is identical to the bioburden method suitability test. Both tests are in place to ensure there is nothing in the test system that will inhibit viable microorganisms from replicating.
Section 6.1.1 of 11737-1:2018, entitled “Selection of an appropriate method,” now provides an additional consideration, which states: “a) neutralization of inhibitory substances, if needed.”
This concept was mentioned in 6.1.2.3 of the 2011 version of the standard, but it was not listed as one of the specific items to address in selection of an appropriate method. In the 2018 version of the standard, it was deemed important enough to add as one of the required items for selection of a test method.
This same requirement was added to 7.2 under validation where it states: “a) assessment of test method suitability to demonstrate lack of inhibition of growth in the test.” Along with this new requirement, additional guidance was included in Annex A, under A.7.2.1.
The additions are specifically written so that a manufacturer might choose to omit performing the bioburden method suitability test if they have a detailed understanding of all components and manufacturing processes relating to their product. The inclusion of words such as “if needed” and “assessment” allow this flexibility. Based on this detailed understanding a manufacturer might know for a fact there are no inhibitory substances on, or in, their product and can provide a written rationale for not performing the test.
#2: Recovery Efficiency
Bioburden testing usually includes an extraction or removal of microorganisms from the product being tested, and that extraction process is rarely perfect in removing 100 percent of the microorganisms. The effectiveness of the bioburden extraction process is determined in a recovery efficiency test. The importance of performing recovery efficiency testing has always been included, and is still represented in the 2018 version, but some details were added and some changes were made.
Section 7.2—Validation—has a new phrase added to the end of sub-section b) that states the recovery efficiency shall be performed “…if appropriate for the purpose for which the data are being generated.”
This additional text enables a manufacturer to determine that recovery efficiency testing might not be necessary in certain circumstances (e.g., performing bioburden testing of product components where only an understanding of incoming bioburden is needed). This addition gives manufacturers more flexibility than what was allowed in the previous version.
Section A.7.2 in Annex A of the previous version underwent a more substantial change. It previously indicated that if the recovery efficiency percentage was less than 50 percent, improvements or alternate techniques should be considered. The 50 percent value was arbitrarily selected and not based on data. Since the use of an arbitrary value is not the best approach, the focus is now on consistency of the results obtained rather than whether a specific value has been achieved. Section A.7.2 no longer contains information on evaluation of recovery efficiency data, but new guidance is provided in Annex C.
Annex C, section C.5.1 states:
The document provides ideas on how a company might select a desired recovery efficiency value and states that for some product types, a recovery efficiency of 20 percent might be appropriate. It is important to note it is not required that companies establish a desired recovery efficiency for each product type.
Information on reviewing recovery efficiency data is provided in sections C.1.3.2 and C.5.1, which state (in order):
Another aspect of recovery efficiency was addressed in the new version regarding the selection of recovery efficiency approach (i.e., repetitive recovery versus inoculated product) in Annex C, section C.1.4 and Table C.1. It suggests that a repetitive recovery efficiency is appropriate for products with a moderate to high bioburden (e.g., 100 CFU and higher), and an inoculated product recovery efficiency is appropriate for products with a low or very low bioburden (e.g., <100 CFU). Although it is not intended that these suggested values are exact cutoff points for either method, they do provide general guidance on when one might be more appropriate than the other.
#3: Limit of Detection
More emphasis is being placed on the limit of detection (LOD) of bioburden tests. Bioburden testing is not meant to be an exact science, such as analytical chemistry, because the bioburden test involves two variables that are sometimes not quantifiable. The first variable is that humans are usually involved in the manufacturing/assembly process, and humans vary in how they interact with the products from day to day. The second variable is that the test is meant to detect living organisms, and organisms vary in how they replicate, remain static, or die due to subtle differences in their environment. Neither variable is consistent in different circumstances, and they are not easily explainable with the laws of physics. Thus, it is not possible to expect bioburden test results to be as accurate or precise as one would with an analytical chemistry test. That being said, however, it is important to take reasonable measures to make bioburden results as valid as possible. Attempting to have a low LOD is one of those reasonable measures to consider.
Section 8.2 states: “The method selected shall take into account factors that will affect the results, such as the limits of detection and plate counting.”
In developing the standard, it was not desired to mandate any particular LOD. The intent of Section 8.2 was to point out the importance of designing bioburden tests with the lowest LOD practicable. An example of this is the debate between a spread plate test method of 1.0 mL (with an LOD of 50) and membrane filtration method of 25 mL (with an LOD of 2). Although the spread plate is faster and easier, if zero colonies are detected on the plate, the results would be reported as less-than 50 CFU (i.e., <50). Whereas with a membrane filtration method of a greater volume the results would be <2 CFU and the data will be more representative of the whole product.
Section A.8.2.1 is a new section that addresses the topic of proper reporting of bioburden values when zero colonies are observed on an agar plate.
Additional guidance is provided in the standard in the form of tables with examples. Having this information, with the example and ideas on improving LODs, is a benefit to the industry. When these concepts are employed, bioburden data can become more useful to manufacturers in detecting changes before they become a problem, in trending manufacturing practices, and in establishing alert and action levels.
#4: Testing of Packaging
The previous version of the standard was missing guidance regarding whether product packaging should be tested for bioburden or not. Since guidance on this topic was excluded from the standard, some manufacturers performed routine bioburden testing for all inner packaging. Some in the industry believed that routine testing of all interior packaging was required (e.g., trays, tray lids, protective packaging, etc.) when the outer package carried the sterile label claim.
In an effort to correct this practice, guidance was added to section A.5.1.1 which states:
“When sampling for the determination of bioburden, a product should be contained in its usual packaging. Typically, it is sufficient to perform a bioburden determination on a product after its removal from its packaging system and to omit the packaging system from the determination. Depending upon the sterile label claim, internal packaging components, such as a tray or product insert, may need to be tested based upon factors such as:
There are two concepts of sterile healthcare products that come into play regarding this new guidance. First is that packaging usually does not have direct contact with the patient, which makes the potential risk to the patient lower for the packaging than for product itself. Some might say that contaminated packaging can transfer microorganisms to product, which then can transfer to the patient. Note the first sentence in A.5.1.1, which states the “usual packaging” should be used for bioburden testing. This means any transfer of microorganisms that could occur on product used on patients will also occur on product used for testing; thus, any microbiological contribution of packaging is accounted for.
Second, bioburden testing of packaging unnecessarily complicates the bioburden test. Inclusion of packaging usually entails additional cutting and manipulation to ensure that it will fit into the container used for testing. Some might say that swabbing could be used to remedy that issue. However issues associated with swabbing such as poor recovery efficiency from the surface to the swab and then poor recovery from the swab to the test system indicate this might not be the best method. Some of the packaging will float on top of the extraction fluid, making a full extraction of the surface area difficult. Also, when packaging is tested with product, it usually fills the container which further increases the difficulty of extraction. One answer is to test packaging separately from the product. This is almost always best practice, but it does add cost to the testing. When there is added cost but little or no true benefit, continued use of the practice should be questioned.
Manufacturers need to understand the potential contribution to product bioburden that packaging can make. Therefore, performing some bioburden testing of packaging to obtain data is a good practice. Manufacturers are usually recommended to test packaging initially, but in a separate container to determine the separate packaging bioburden counts from product bioburden counts. Once the data are gathered and it is determined that the results are acceptable, there is no need to test packaging on a routine basis. At that point, proper implementation of change control and good microbiological controls in inspection and storage processes is sufficient.
#5: Table of Responsibilities
Although compliance with device standards is ultimately the responsibility of the manufacturer, there has been confusion in the industry regarding where some responsibilities lay. In an attempt to provide clearer guidance for both manufacturers and testing laboratories, a table of responsibilities has been included in Annex D. This table indicates where collaborative efforts between the manufacturer and the testing lab would ensure the best testing method is utilized for the product in question.
The table provides guidance for most of the sections found in the standard and uses the abbreviations R = responsibility, I = this can involve providing assistance or information, and N/A = not generally applicable. Examples of where it is the responsibility of both the manufacturer and laboratory include selection of bioburden method, test method suitability for validation of the bioburden method, and removal technique. Whereas, specification of acceptable bioburden levels and trending are the responsibility of the manufacturer and items such as preparation and sterilization of materials and microbial characterization are laboratory responsibilities. In most cases, assistance and information from both sides is the best approach.
Correction to the Standard
While items are the top five changes in the new revision deemed important enough to highlight, there is one additional item that needs to be mentioned. A formula that may be used for the MPN test was added to Annex B.3.3.4. The formula placed into the standard is incorrect and is in the process of being corrected. The proper formula should be:
MPN = Ln(number tested/number negative for growth) ×1/SIP
This is a simplified version of the original formula provided by Cochran in 19501, which was:
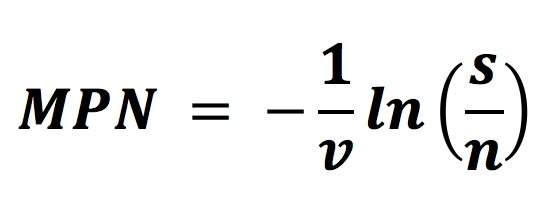
The MPN method is beneficial when applied correctly.
Conclusion
Bioburden is one of the most common tests performed in the medtech industry and is a valuable contributor to demonstrating long-term control of manufacturing sites. One of the goals of this revision of the bioburden standard was to provide additional guidance to manufacturers and laboratories to improve the quality of bioburden testing and data reporting. A detailed understanding of the concepts discussed in this article will help manufacturers to ensure bioburden tests are appropriate for the products being tested and the responsibilities stated in the standard have been correctly addressed.
Reference
1 Cochran, W. “Estimation of Bacterial Densities by Means of the Most Probable Number,” Biometrics. 6:105-116, 1950.
Martell K. Winters, B.S., SM (NRCM), director of scientific competency at Nelson Laboratories, is a recognized authority and industry leader on radiation sterilization, bioburden, microbiology, aseptic processing, environmental control, and tissue processing and disinfection. He has over 20 years of experience working in research, laboratory, test design, and manufacturing microbiology functions in the medical device, tissue, and pharmaceutical industries. Winters has worked with clients in all areas of the world to speak, train, or consult on failure investigations, process design, process controls, sterilization, and aseptic processing. He is an active committee member of many working groups within the Association for the Advancement of Medical Instrumentation (AAMI), and with the American Association of Tissue Banks (AATB) and the International Organization for Standardization (ISO).
Wendy Wangsgard, Ph.D., RM (NRCM), senior scientist at Nelson Laboratories, is a recognized authority and industry leader on radiation sterilization and bioburden. She has over 17 years of experience working in research and laboratory functions in research, medical device, and tissue industries. Wendy has worked with clients to consult on failure investigations, bioburden issues, establishing bioburden alert and action levels, product family grouping, radiation sterilization, and tissue validations. She is a member of the Association for the Advancement of Medical Instrumentation (AAMI) involved in the working groups for Radiation Sterilization, Microbiological Methods, Assurance of Sterility, Sterilization Terminology and the Compatibility of Materials Subject to Sterilization.
#1: Bioburden Method Suitability
Bioburden test methods are dependent on the ability of microorganisms to replicate in the bioburden test system. Some products tested for bioburden can release substances that inhibit microorganism replication. The inhibitory substance is typically known to the manufacturer because it is intentionally included as part of the product (i.e., chemical or antibiotic). Sometimes, however, an unknown inhibitory substance can be present that can be problematic because the manufacturer may believe there is no need to test for inhibitory substances. For example, there might be residuals from cleaning or disinfecting processes on the product, or on components of the product that are provided by a supplier.
When an inhibitory substance is present, the bioburden test results can look very low (e.g., 0 CFU), even though there are viable microorganisms on the product. This is because inhibitory substances sometimes do not inactivate or kill the microorganisms; they merely inhibit them from replicating.
There is an analogous test used in qualifying a test of sterility. It has historically been called the Bacteriostasis/Fungistasis test or B/F test; it is currently called Method Suitability. The intent behind that test is identical to the bioburden method suitability test. Both tests are in place to ensure there is nothing in the test system that will inhibit viable microorganisms from replicating.
Section 6.1.1 of 11737-1:2018, entitled “Selection of an appropriate method,” now provides an additional consideration, which states: “a) neutralization of inhibitory substances, if needed.”
This concept was mentioned in 6.1.2.3 of the 2011 version of the standard, but it was not listed as one of the specific items to address in selection of an appropriate method. In the 2018 version of the standard, it was deemed important enough to add as one of the required items for selection of a test method.
This same requirement was added to 7.2 under validation where it states: “a) assessment of test method suitability to demonstrate lack of inhibition of growth in the test.” Along with this new requirement, additional guidance was included in Annex A, under A.7.2.1.
The additions are specifically written so that a manufacturer might choose to omit performing the bioburden method suitability test if they have a detailed understanding of all components and manufacturing processes relating to their product. The inclusion of words such as “if needed” and “assessment” allow this flexibility. Based on this detailed understanding a manufacturer might know for a fact there are no inhibitory substances on, or in, their product and can provide a written rationale for not performing the test.
#2: Recovery Efficiency
Bioburden testing usually includes an extraction or removal of microorganisms from the product being tested, and that extraction process is rarely perfect in removing 100 percent of the microorganisms. The effectiveness of the bioburden extraction process is determined in a recovery efficiency test. The importance of performing recovery efficiency testing has always been included, and is still represented in the 2018 version, but some details were added and some changes were made.
Section 7.2—Validation—has a new phrase added to the end of sub-section b) that states the recovery efficiency shall be performed “…if appropriate for the purpose for which the data are being generated.”
This additional text enables a manufacturer to determine that recovery efficiency testing might not be necessary in certain circumstances (e.g., performing bioburden testing of product components where only an understanding of incoming bioburden is needed). This addition gives manufacturers more flexibility than what was allowed in the previous version.
Section A.7.2 in Annex A of the previous version underwent a more substantial change. It previously indicated that if the recovery efficiency percentage was less than 50 percent, improvements or alternate techniques should be considered. The 50 percent value was arbitrarily selected and not based on data. Since the use of an arbitrary value is not the best approach, the focus is now on consistency of the results obtained rather than whether a specific value has been achieved. Section A.7.2 no longer contains information on evaluation of recovery efficiency data, but new guidance is provided in Annex C.
Annex C, section C.5.1 states:
-
“Due to the variability of design, materials, product configurations, manufacturing processes, etc., it is not required by this document that a particular bioburden recovery efficiency result be obtained. However, if bioburden recovery efficiency results fall below a target or desired value, another technique should be attempted (e.g. addition of another extraction method or lengthening the current extraction method) to determine if better results can be obtained.”
The document provides ideas on how a company might select a desired recovery efficiency value and states that for some product types, a recovery efficiency of 20 percent might be appropriate. It is important to note it is not required that companies establish a desired recovery efficiency for each product type.
Information on reviewing recovery efficiency data is provided in sections C.1.3.2 and C.5.1, which state (in order):
-
“When reviewing bioburden recovery efficiency results, a review of the consistency of results, or lack thereof, can indicate that a different extraction method should be applied.”
“Nevertheless, unexpectedly low or widely distributed bioburden recovery efficiency might not be appropriate depending on the criticality and purpose of the bioburden data, and, if this is the case, further improvement of the removal technique (e.g. enhanced by disassembling, more intensive mechanical shaking, active rinsing of cavities, prolongation of rinsing time, modification of eluent) should be investigated.”
Another aspect of recovery efficiency was addressed in the new version regarding the selection of recovery efficiency approach (i.e., repetitive recovery versus inoculated product) in Annex C, section C.1.4 and Table C.1. It suggests that a repetitive recovery efficiency is appropriate for products with a moderate to high bioburden (e.g., 100 CFU and higher), and an inoculated product recovery efficiency is appropriate for products with a low or very low bioburden (e.g., <100 CFU). Although it is not intended that these suggested values are exact cutoff points for either method, they do provide general guidance on when one might be more appropriate than the other.
#3: Limit of Detection
More emphasis is being placed on the limit of detection (LOD) of bioburden tests. Bioburden testing is not meant to be an exact science, such as analytical chemistry, because the bioburden test involves two variables that are sometimes not quantifiable. The first variable is that humans are usually involved in the manufacturing/assembly process, and humans vary in how they interact with the products from day to day. The second variable is that the test is meant to detect living organisms, and organisms vary in how they replicate, remain static, or die due to subtle differences in their environment. Neither variable is consistent in different circumstances, and they are not easily explainable with the laws of physics. Thus, it is not possible to expect bioburden test results to be as accurate or precise as one would with an analytical chemistry test. That being said, however, it is important to take reasonable measures to make bioburden results as valid as possible. Attempting to have a low LOD is one of those reasonable measures to consider.
Section 8.2 states: “The method selected shall take into account factors that will affect the results, such as the limits of detection and plate counting.”
In developing the standard, it was not desired to mandate any particular LOD. The intent of Section 8.2 was to point out the importance of designing bioburden tests with the lowest LOD practicable. An example of this is the debate between a spread plate test method of 1.0 mL (with an LOD of 50) and membrane filtration method of 25 mL (with an LOD of 2). Although the spread plate is faster and easier, if zero colonies are detected on the plate, the results would be reported as less-than 50 CFU (i.e., <50). Whereas with a membrane filtration method of a greater volume the results would be <2 CFU and the data will be more representative of the whole product.
Section A.8.2.1 is a new section that addresses the topic of proper reporting of bioburden values when zero colonies are observed on an agar plate.
-
“Limits of detection (LOD) for bioburden test methods should be taken into account in determining the bioburden value. For microbiology reporting, when a portion of the extract is tested for bioburden, and zero colonies are recovered, the results are typically reported as less than ‘X’ where ‘1/X’ is representative of the fraction of the portion tested. For example, if a product is extracted in 400 ml and 1/4 of the extract is filtered, results of zero colonies will be reported as less than 4 colony forming units (i.e. <4 CFU). Therefore, the LOD for this example is 4. In microbiological reporting, a result of <4 CFU means that it is possible that the entire extract contains either 0, 1, 2 or 3 CFUs, but microbiological reporting rules require that it be reported as <4 CFU. Individual bioburden results are reported in whole numbers because the number is representative of a colony forming unit. Averages or other mathematical calculations using bioburden data are typically reported to one decimal place. LOD can be improved by the following:
a) modification to the test method (e.g., filtering a larger portion of the extract);
b) pooling multiple samples;
c) utilizing another test method, such as MPN.”
Additional guidance is provided in the standard in the form of tables with examples. Having this information, with the example and ideas on improving LODs, is a benefit to the industry. When these concepts are employed, bioburden data can become more useful to manufacturers in detecting changes before they become a problem, in trending manufacturing practices, and in establishing alert and action levels.
#4: Testing of Packaging
The previous version of the standard was missing guidance regarding whether product packaging should be tested for bioburden or not. Since guidance on this topic was excluded from the standard, some manufacturers performed routine bioburden testing for all inner packaging. Some in the industry believed that routine testing of all interior packaging was required (e.g., trays, tray lids, protective packaging, etc.) when the outer package carried the sterile label claim.
In an effort to correct this practice, guidance was added to section A.5.1.1 which states:
“When sampling for the determination of bioburden, a product should be contained in its usual packaging. Typically, it is sufficient to perform a bioburden determination on a product after its removal from its packaging system and to omit the packaging system from the determination. Depending upon the sterile label claim, internal packaging components, such as a tray or product insert, may need to be tested based upon factors such as:
- What is intended to be sterile,
- When the package is an integral part of the product, or
- For specific evaluation.”
There are two concepts of sterile healthcare products that come into play regarding this new guidance. First is that packaging usually does not have direct contact with the patient, which makes the potential risk to the patient lower for the packaging than for product itself. Some might say that contaminated packaging can transfer microorganisms to product, which then can transfer to the patient. Note the first sentence in A.5.1.1, which states the “usual packaging” should be used for bioburden testing. This means any transfer of microorganisms that could occur on product used on patients will also occur on product used for testing; thus, any microbiological contribution of packaging is accounted for.
Second, bioburden testing of packaging unnecessarily complicates the bioburden test. Inclusion of packaging usually entails additional cutting and manipulation to ensure that it will fit into the container used for testing. Some might say that swabbing could be used to remedy that issue. However issues associated with swabbing such as poor recovery efficiency from the surface to the swab and then poor recovery from the swab to the test system indicate this might not be the best method. Some of the packaging will float on top of the extraction fluid, making a full extraction of the surface area difficult. Also, when packaging is tested with product, it usually fills the container which further increases the difficulty of extraction. One answer is to test packaging separately from the product. This is almost always best practice, but it does add cost to the testing. When there is added cost but little or no true benefit, continued use of the practice should be questioned.
Manufacturers need to understand the potential contribution to product bioburden that packaging can make. Therefore, performing some bioburden testing of packaging to obtain data is a good practice. Manufacturers are usually recommended to test packaging initially, but in a separate container to determine the separate packaging bioburden counts from product bioburden counts. Once the data are gathered and it is determined that the results are acceptable, there is no need to test packaging on a routine basis. At that point, proper implementation of change control and good microbiological controls in inspection and storage processes is sufficient.
#5: Table of Responsibilities
Although compliance with device standards is ultimately the responsibility of the manufacturer, there has been confusion in the industry regarding where some responsibilities lay. In an attempt to provide clearer guidance for both manufacturers and testing laboratories, a table of responsibilities has been included in Annex D. This table indicates where collaborative efforts between the manufacturer and the testing lab would ensure the best testing method is utilized for the product in question.
The table provides guidance for most of the sections found in the standard and uses the abbreviations R = responsibility, I = this can involve providing assistance or information, and N/A = not generally applicable. Examples of where it is the responsibility of both the manufacturer and laboratory include selection of bioburden method, test method suitability for validation of the bioburden method, and removal technique. Whereas, specification of acceptable bioburden levels and trending are the responsibility of the manufacturer and items such as preparation and sterilization of materials and microbial characterization are laboratory responsibilities. In most cases, assistance and information from both sides is the best approach.
Correction to the Standard
While items are the top five changes in the new revision deemed important enough to highlight, there is one additional item that needs to be mentioned. A formula that may be used for the MPN test was added to Annex B.3.3.4. The formula placed into the standard is incorrect and is in the process of being corrected. The proper formula should be:
MPN = Ln(number tested/number negative for growth) ×1/SIP
This is a simplified version of the original formula provided by Cochran in 19501, which was:
The MPN method is beneficial when applied correctly.
Conclusion
Bioburden is one of the most common tests performed in the medtech industry and is a valuable contributor to demonstrating long-term control of manufacturing sites. One of the goals of this revision of the bioburden standard was to provide additional guidance to manufacturers and laboratories to improve the quality of bioburden testing and data reporting. A detailed understanding of the concepts discussed in this article will help manufacturers to ensure bioburden tests are appropriate for the products being tested and the responsibilities stated in the standard have been correctly addressed.
Reference
1 Cochran, W. “Estimation of Bacterial Densities by Means of the Most Probable Number,” Biometrics. 6:105-116, 1950.
Martell K. Winters, B.S., SM (NRCM), director of scientific competency at Nelson Laboratories, is a recognized authority and industry leader on radiation sterilization, bioburden, microbiology, aseptic processing, environmental control, and tissue processing and disinfection. He has over 20 years of experience working in research, laboratory, test design, and manufacturing microbiology functions in the medical device, tissue, and pharmaceutical industries. Winters has worked with clients in all areas of the world to speak, train, or consult on failure investigations, process design, process controls, sterilization, and aseptic processing. He is an active committee member of many working groups within the Association for the Advancement of Medical Instrumentation (AAMI), and with the American Association of Tissue Banks (AATB) and the International Organization for Standardization (ISO).
Wendy Wangsgard, Ph.D., RM (NRCM), senior scientist at Nelson Laboratories, is a recognized authority and industry leader on radiation sterilization and bioburden. She has over 17 years of experience working in research and laboratory functions in research, medical device, and tissue industries. Wendy has worked with clients to consult on failure investigations, bioburden issues, establishing bioburden alert and action levels, product family grouping, radiation sterilization, and tissue validations. She is a member of the Association for the Advancement of Medical Instrumentation (AAMI) involved in the working groups for Radiation Sterilization, Microbiological Methods, Assurance of Sterility, Sterilization Terminology and the Compatibility of Materials Subject to Sterilization.