U.S. Food and Drug Administration12.13.19
The U.S. Food and Drug Administration authorized marketing of the first test to aid in newborn screening for Duchenne Muscular Dystrophy (DMD), a rare genetic disorder that causes progressive muscle deterioration and weakness.
“Diagnostics that can safely and effectively screen newborns can help health care professionals identify and discuss potential treatment options with parents and caregivers before symptoms or effects on a baby’s health may be noticeable,” said Tim Stenzel, M.D., Ph.D., director of the Office of In Vitro Diagnostics and Radiological Health in the FDA’s Center for Devices and Radiological Health. “This authorization reflects our commitment to fostering innovation in devices to help inform and provide options to patients and their caregivers. Early screening can help identify individuals who need additional follow up or treatment.”
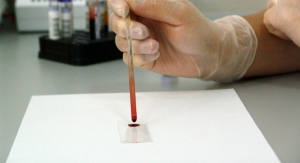
The GSP Neonatal Creatine Kinase-MM kit authorized is intended to aid in screening newborns for DMD. Newborn screening is a series of tests to help health care professionals identify serious diseases and conditions shortly after birth. As part of this screening, a newborn screening card is used to collect a small amount of blood from a prick of an infant’s heel, sometimes called a heel stick. The collected, dried blood samples are used to test for a variety of diseases and conditions.
While the number and type of diseases and conditions tested on each state’s newborn screening panel can vary, there has been a national effort to harmonize screening practices across state newborn screening programs in the U.S. As a result of the collaboration between the federal Advisory Committee on Heritable Disorders in Newborns and Children and the American College of Medical Genetics, as well as governmental, non-governmental, advocacy and private partners, the Recommended Uniform Screening Panel (RUSP) was developed and adopted. The RUSP is a list of core and secondary conditions for screening newborns that the U.S. Department of Health and Human Services recommends for states to screen as part of their state universal newborn screening programs.
This authorization of the GSP Neonatal Creatine Kinase-MM kit enables laboratories to add this test to their newborn screening panel if they choose to do so, but this authorization does not signal a recommendation for DMD to be added to the RUSP as a condition for which newborn screening is recommended. The GSP Neonatal Creatine Kinase-MM kit is not intended for DMD diagnosis or for screening of other forms of muscular dystrophies.
The GSP Neonatal Creatine Kinase-MM kit works by measuring the concentration of a type of protein called CK-MM, which is part of a group of proteins called creatine kinase. Creatine kinase is found in muscle tissue and CK-MM enters the blood stream in increased amounts when there is muscle damage. This test measures the levels of CK-MM from the dried blood samples collected from the prick of a newborn’s heel 24 to 48 hours after birth. Elevated levels of CK-MM detected by the kit may indicate presence of DMD. Results showing elevated CK-MM must be confirmed using other testing methods, such as muscle biopsies, genetic and other laboratory tests.
DMD, while rare, is the most common type of muscular dystrophy. It is caused by an absence of dystrophin, a protein that helps keep muscle cells intact. The first symptoms are usually seen between 3 and 5 years of age and worsen over time. The disease often occurs in people without a known family history of the condition and primarily affects boys, but in rare cases it can affect girls. DMD occurs in about 1 in 3,600 male live-born infants worldwide.
People with DMD progressively lose the ability to perform activities independently and often require use of a wheelchair by their early teens. As the disease progresses, life-threatening heart and respiratory conditions can occur. Patients typically succumb to the disease in their 20s or 30s; however, disease severity and life expectancy vary. The U.S. Centers for Disease Control and Prevention (CDC) advises that early diagnosis could lead to more personalized care for each person living with muscular dystrophy and may give each of them a better chance to reach his or her full potential. The CDC has also found that there was an average of two and a half years between when a parent or caregiver noticed the first signs and symptoms of DMD, and when a diagnosis of DMD was made.
The FDA reviewed the GSP Neonatal Creatine Kinase-MM kit through the de novo premarket review pathway, a regulatory pathway for low-to-moderate risk devices of a new type. During this process, the FDA evaluated data from a clinical study of 3,041 newborns whose dried blood samples were tested for protein levels that are associated with DMD. In the study, the kit was able to accurately identify the four screened newborns that had DMD-causing genetic mutations. The device manufacturer also tested 30 samples from newborns with clinically confirmed cases of DMD, all of which were correctly identified by the test.
Along with this authorization, the FDA is establishing criteria, called special controls, that must be met for tests of this type, including certain design verification, design validation and labeling requirements. When met, these special controls, along with general controls, provide a reasonable assurance of safety and effectiveness for tests of this type. This action also creates a new regulatory classification, which means that subsequent devices of the same type, with the same intended use, may go through the FDA’s 510(k) pathway, whereby devices can obtain clearance by demonstrating substantial equivalence to a predicate device.
Risks associated with use of the kit include false negative test results. As part of the clinical study, the device manufacturer performed genetic testing, an accepted method of diagnosing DMD, on 173 patient samples including a subset of patients identified as negative by the GSP Neonatal Creatine Kinase-MM kit. Genetic testing on the negative samples did not identify any DMD-causing genetic variants, confirming the negative screening results by the GSP Neonatal Creatine Kinase-MM kit.
The FDA granted marketing authorization of the GSP Neonatal Creatine Kinase-MM kit to PerkinElmer.
“Diagnostics that can safely and effectively screen newborns can help health care professionals identify and discuss potential treatment options with parents and caregivers before symptoms or effects on a baby’s health may be noticeable,” said Tim Stenzel, M.D., Ph.D., director of the Office of In Vitro Diagnostics and Radiological Health in the FDA’s Center for Devices and Radiological Health. “This authorization reflects our commitment to fostering innovation in devices to help inform and provide options to patients and their caregivers. Early screening can help identify individuals who need additional follow up or treatment.”
The GSP Neonatal Creatine Kinase-MM kit authorized is intended to aid in screening newborns for DMD. Newborn screening is a series of tests to help health care professionals identify serious diseases and conditions shortly after birth. As part of this screening, a newborn screening card is used to collect a small amount of blood from a prick of an infant’s heel, sometimes called a heel stick. The collected, dried blood samples are used to test for a variety of diseases and conditions.
While the number and type of diseases and conditions tested on each state’s newborn screening panel can vary, there has been a national effort to harmonize screening practices across state newborn screening programs in the U.S. As a result of the collaboration between the federal Advisory Committee on Heritable Disorders in Newborns and Children and the American College of Medical Genetics, as well as governmental, non-governmental, advocacy and private partners, the Recommended Uniform Screening Panel (RUSP) was developed and adopted. The RUSP is a list of core and secondary conditions for screening newborns that the U.S. Department of Health and Human Services recommends for states to screen as part of their state universal newborn screening programs.
This authorization of the GSP Neonatal Creatine Kinase-MM kit enables laboratories to add this test to their newborn screening panel if they choose to do so, but this authorization does not signal a recommendation for DMD to be added to the RUSP as a condition for which newborn screening is recommended. The GSP Neonatal Creatine Kinase-MM kit is not intended for DMD diagnosis or for screening of other forms of muscular dystrophies.
The GSP Neonatal Creatine Kinase-MM kit works by measuring the concentration of a type of protein called CK-MM, which is part of a group of proteins called creatine kinase. Creatine kinase is found in muscle tissue and CK-MM enters the blood stream in increased amounts when there is muscle damage. This test measures the levels of CK-MM from the dried blood samples collected from the prick of a newborn’s heel 24 to 48 hours after birth. Elevated levels of CK-MM detected by the kit may indicate presence of DMD. Results showing elevated CK-MM must be confirmed using other testing methods, such as muscle biopsies, genetic and other laboratory tests.
DMD, while rare, is the most common type of muscular dystrophy. It is caused by an absence of dystrophin, a protein that helps keep muscle cells intact. The first symptoms are usually seen between 3 and 5 years of age and worsen over time. The disease often occurs in people without a known family history of the condition and primarily affects boys, but in rare cases it can affect girls. DMD occurs in about 1 in 3,600 male live-born infants worldwide.
People with DMD progressively lose the ability to perform activities independently and often require use of a wheelchair by their early teens. As the disease progresses, life-threatening heart and respiratory conditions can occur. Patients typically succumb to the disease in their 20s or 30s; however, disease severity and life expectancy vary. The U.S. Centers for Disease Control and Prevention (CDC) advises that early diagnosis could lead to more personalized care for each person living with muscular dystrophy and may give each of them a better chance to reach his or her full potential. The CDC has also found that there was an average of two and a half years between when a parent or caregiver noticed the first signs and symptoms of DMD, and when a diagnosis of DMD was made.
The FDA reviewed the GSP Neonatal Creatine Kinase-MM kit through the de novo premarket review pathway, a regulatory pathway for low-to-moderate risk devices of a new type. During this process, the FDA evaluated data from a clinical study of 3,041 newborns whose dried blood samples were tested for protein levels that are associated with DMD. In the study, the kit was able to accurately identify the four screened newborns that had DMD-causing genetic mutations. The device manufacturer also tested 30 samples from newborns with clinically confirmed cases of DMD, all of which were correctly identified by the test.
Along with this authorization, the FDA is establishing criteria, called special controls, that must be met for tests of this type, including certain design verification, design validation and labeling requirements. When met, these special controls, along with general controls, provide a reasonable assurance of safety and effectiveness for tests of this type. This action also creates a new regulatory classification, which means that subsequent devices of the same type, with the same intended use, may go through the FDA’s 510(k) pathway, whereby devices can obtain clearance by demonstrating substantial equivalence to a predicate device.
Risks associated with use of the kit include false negative test results. As part of the clinical study, the device manufacturer performed genetic testing, an accepted method of diagnosing DMD, on 173 patient samples including a subset of patients identified as negative by the GSP Neonatal Creatine Kinase-MM kit. Genetic testing on the negative samples did not identify any DMD-causing genetic variants, confirming the negative screening results by the GSP Neonatal Creatine Kinase-MM kit.
The FDA granted marketing authorization of the GSP Neonatal Creatine Kinase-MM kit to PerkinElmer.