Ranica Arrowsmith, Associate Editor03.13.14
“I’m afraid I have no news to break although a lot is going on behind the scenes,” said Serge Bernasconi, CEO of MedTech Europe, the European Diagnostic Manufacturers Association (EDMA) and Eucomed, of the ongoing proposed revisions to the European Union’s Medical Devices (MDD) and In Vitro Diagnostics Directives (IVDD). Bernasconi’s Feb. 26 update outlined continued concerns with the proposed changes, but no concrete updates.
According to a January position paper released by EDMA, while the European Commission (EC) has proposed a transition period of five years for in-vitro diagnostics (IVDs) to adapt to changes in the law, the European parliament voted for a transition of only three years. EDMA maintains that it strongly supports a five-year transition period for IVDs.
“This timeframe is needed for manufacturers to be able to fully comply with the various new requirements and place all of the necessary manufacturing processes in place,” the paper said. “The three-year time period proposed by the European parliament is simply not realistically achievable.”
Subsequently, Bernasconi reported that the three-year transition period “continues to worry” the coalition of medtech associations.
“Three years is simply not enough time for all involved players to adapt to what are really sweeping changes across the system,” he said. “We’re not talking only manufacturers (and importers) but also notified bodies, competent authorities, reference laboratories and the European Commission who will need to manage the changes in three years. Implementing changes over five years is tough, but it is a more feasible scenario, one that all involved players can fulfill with effort and dedication. I believe that if we go for a three-year transition period we will probably encounter a request for an extension after two and a half years in the process.”
Nonetheless, speaking for industry, Bernasconi noted that the many of the proposed changes to the MDD and IVDD are welcome, as they will improve global competitiveness of the European IVD industry and facilitate access to new technology for patients. One of the key changes would be implementing a classification model that is harmonized with international standards, and Bernasconi said that this would set the scene for comparing relevant data on a global level and improving vigilance.
“This is a critical aspect for enhancing patient safety and one that should, after all, be at the core of the legislative changes,” Bernasconi said.
Several MDD-related issues emerged as most important over the past months of negotiation: First, the industry associations claim the commission’s proposal for the re-processing of single-use medical devices is reasonable; the associations also are keen to see a more balanced approach on clinical requirements, but not if it means simply applying pharmaceutical requirements to devices; and phasing out “hazardous substances” in medical devices is supported by the industry, but only if companies are permitted to phase them out over a fair timeline rather than immediately.
Finally, Bernasconi noted that the EC’s proposed scrutiny procedure is not very clear (see figure 1).
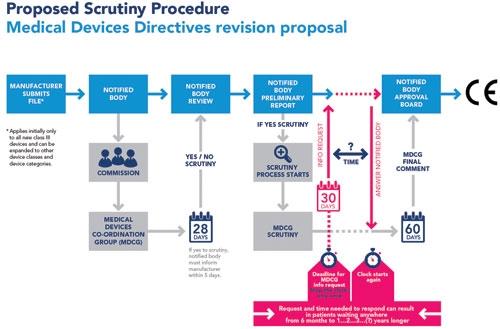
Image courtesy of Eucomed.
“Lately there’s been some confusion on exactly what industry proposed as an alternative,” Bernasconi said. “…We find the commission procedure to be a random, ‘needle-in-a-haystack’ approach—the system should not be ‘looking for a needle in a haystack’ through random checks at the end of a review process but should prevent the needle landing in the haystack in the first place … we want to reinforce government control of the system—to empower authorities to keep closer tabs on notified bodies and manufacturers through tools like unannounced visits. At some point though, our ‘systematic control procedure’ became known in MDD circles as ‘systematic scrutiny,’ leading people to believe that industry would support some version of the commission’s proposed scrutiny procedure. I want it to be clear that this is not the case. So as of today, I’m renaming our alternative. What industry wants is a ‘reinforced control procedure.’”
Last year, Eucomed released a position paper to counter the EC’s latest efforts at overhauling the MDD and IVDD. In it, the association stressed that it supports changes that improve patient safety, do not unnecessary delay patient access to medical devices that save or improve lives and do not hamper innovation.
However, Eucomed also laid out seven key focal areas that it wanted to see before it could give the commission its full support:
It seems that until the industry and European authorities find some middle ground, any progress toward MDD and IVDD renovation might be slow-going.
According to a January position paper released by EDMA, while the European Commission (EC) has proposed a transition period of five years for in-vitro diagnostics (IVDs) to adapt to changes in the law, the European parliament voted for a transition of only three years. EDMA maintains that it strongly supports a five-year transition period for IVDs.
“This timeframe is needed for manufacturers to be able to fully comply with the various new requirements and place all of the necessary manufacturing processes in place,” the paper said. “The three-year time period proposed by the European parliament is simply not realistically achievable.”
Subsequently, Bernasconi reported that the three-year transition period “continues to worry” the coalition of medtech associations.
“Three years is simply not enough time for all involved players to adapt to what are really sweeping changes across the system,” he said. “We’re not talking only manufacturers (and importers) but also notified bodies, competent authorities, reference laboratories and the European Commission who will need to manage the changes in three years. Implementing changes over five years is tough, but it is a more feasible scenario, one that all involved players can fulfill with effort and dedication. I believe that if we go for a three-year transition period we will probably encounter a request for an extension after two and a half years in the process.”
Nonetheless, speaking for industry, Bernasconi noted that the many of the proposed changes to the MDD and IVDD are welcome, as they will improve global competitiveness of the European IVD industry and facilitate access to new technology for patients. One of the key changes would be implementing a classification model that is harmonized with international standards, and Bernasconi said that this would set the scene for comparing relevant data on a global level and improving vigilance.
“This is a critical aspect for enhancing patient safety and one that should, after all, be at the core of the legislative changes,” Bernasconi said.
Several MDD-related issues emerged as most important over the past months of negotiation: First, the industry associations claim the commission’s proposal for the re-processing of single-use medical devices is reasonable; the associations also are keen to see a more balanced approach on clinical requirements, but not if it means simply applying pharmaceutical requirements to devices; and phasing out “hazardous substances” in medical devices is supported by the industry, but only if companies are permitted to phase them out over a fair timeline rather than immediately.
Finally, Bernasconi noted that the EC’s proposed scrutiny procedure is not very clear (see figure 1).
Image courtesy of Eucomed.
Last year, Eucomed released a position paper to counter the EC’s latest efforts at overhauling the MDD and IVDD. In it, the association stressed that it supports changes that improve patient safety, do not unnecessary delay patient access to medical devices that save or improve lives and do not hamper innovation.
However, Eucomed also laid out seven key focal areas that it wanted to see before it could give the commission its full support:
- Only the best notified bodies should be allowed to approve medical devices to the market in order to ensure that the backbone of Europe’s decentralized system meets the highest safety and quality standards.
- A systematic control procedure is necessary to improve the system and increase patient safety. The proposed “commission scrutiny procedure” (article 44) is inappropriate because it is not systematic and will not lead to increased patient safety. It should be replaced with a systematic control procedure that goes beyond the current proposed measures. Only then will the EC reach the outcome that is desired by all stakeholders: maximum safety for all Europeans without unnecessary delay or duplication of work.
- Increase stakeholder involvement to ensure the opinions of essential healthcare actors are heard.
- Greater transparency and traceability is critical to ensure that patients, doctors, industry and other stakeholders have access to clear information about the medical devices they use.
- Clinical evidence needs more clarity as clear, appropriate requirements for clinical evidence are paramount to demonstrate that devices perform well and are safe for patients when used by a well-trained healthcare professional and as intended by the manufacturer.
- Enhance vigilance and market surveillance to allow for rapid identification of adverse events and to ensure coherent and timely action by member states.
- Clear, science-based classifications are needed to avoid the currently proposed arbitrary reclassification of families of medical devices without any scientific or other justification, which will lead to global confusion. Clear and science-based procedures must be followed to ensure that devices are appropriately classified.
It seems that until the industry and European authorities find some middle ground, any progress toward MDD and IVDD renovation might be slow-going.