James A. Dunning, QPC Services LLC09.01.17
The medtech and pharmaceutical sectors are both essential to health, and while they have long maintained a symbiotic relationship, the two have never really understood each other. Certainly, there are some common threads connecting the pair—both industries are regulated by the U.S. Food and Drug Administration (FDA), many of their products are available only by prescription, and both are committed to improving patient health and wellness.
There are differences, too: Research and development, brand marketing, product design (mechanical vs. chemical), and technological innovation, to name just a few. Perhaps the most striking disparity, however, exists in FDA regulations, though I contend there are more similarities between the two industries than meets the eye. Hence my motivation for this ongoing series comparing parts of the drug and device industries’ Current Good Manufacturing Practices (cGMP) requirements.
This month’s column will focus on ways 21 CFR Part 820 Quality System Regulation, also known as the cGMP regulation for medical devices, aligns with the cGMP requirements of 21 CFR Part 211 Subpart B—Organization and Personnel. The two most recent articles dealt with Subpart A—General Provisions. As was the case in previous columns, the term “pharmaceutical(s)” is used rather than “drug(s)” unless the term is part of an excerpt from the cGMP regulation. Scope and definition are the same as well.
An overview of the scope of this article is provided in Table 1 (below).
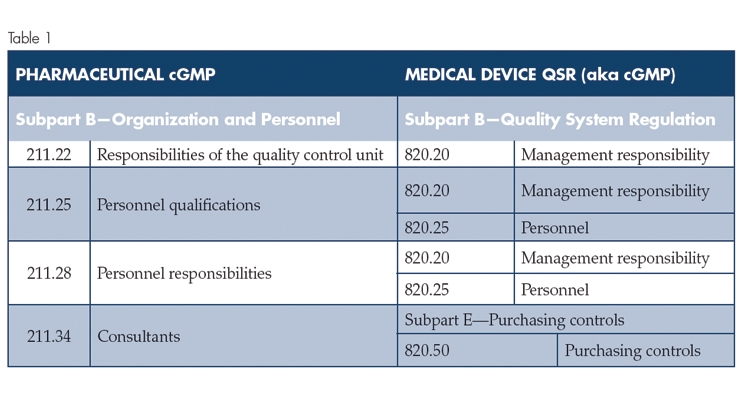
From a high-level overview, the table shows that Medical Device cGMP Subpart B—Quality System Regulation aligns relatively well with Pharmaceutical cGMP Subpart B Organization and Personnel, with the exception of 211.34 Consultants. The medical device cGMP addresses consultants in Subpart E—Purchasing controls. Additionally, the pharmaceutical cGMP does not address quality audits, whereas the Medical Device cGMP Subpart B—Quality System Regulation, 820.22 Quality audit, specifically refers to quality audits. In practice, quality audits are a key Quality Control Unit responsibility, and the expectations for quality auditing are contained in U.S. Food and Drug Administration (FDA) guidance documents.
Pharmaceutical cGMP Requirements, Subpart B—Organization and Personnel, 211.22 (a) declares, “There shall be a quality control unit that shall have the responsibility and authority to approve or reject all components, drug product containers, closures, in-process materials, packaging material, labeling, and drug products, and the authority to review production records to assure that no errors have occurred or, if errors have occurred, that they have been fully investigated. The quality control unit shall be responsible for approving or rejecting drug products manufactured, processed, packed, or held under contract by another company.”
Medical Device cGMP Requirements, Subpart B Quality System Regulations, 820.20 in part, states the following:
“(b) Organization. Each manufacturer shall establish and maintain an adequate organizational structure to ensure that devices are designed and produced in accordance with the requirements of this part. (1) Responsibility and authority. Each manufacturer shall establish the appropriate responsibility, authority, and interrelation of all personnel who manage, perform, and assess work affecting quality, and provide the independence and authority necessary to perform these tasks. (2) Resources. Each manufacturer shall provide adequate resources, including the assignment of trained personnel, for management, performance of work, and assessment activities, including internal quality audits, to meet the requirements of this part. (3) Management representative. Management with executive responsibility shall appoint, and document such appointment of, a member of management who, irrespective of other responsibilities, shall have established authority over and responsibility for: (i) Ensuring that quality system requirements are effectively established and effectively maintained in accordance with this part; and (ii) Reporting on the performance of the quality system to management with executive responsibility for review.”
The approach is different, but the results are similar. I believe the pharmaceutical cGMP mandates more clearly establish the Quality Control Unit requirements, and the directives are more specific, with the exception of the management representative role. In practice, pharmaceutical companies have management representatives.
Personnel qualifications and personnel responsibilities are equally addressed in both the pharmaceutical and medical device cGMP requirements.
For personnel qualifications, Pharmaceutical cGMP 211.25 states in part, “(b) Each person responsible for supervising the manufacture, processing, packing, or holding of a drug product shall have the education, training, and experience, or any combination thereof, to perform assigned functions in such a manner as to provide assurance that the drug product has the safety, identity, strength, quality, and purity that it purports or is represented to possess.” The corresponding prerequisites under Medical Device cGMP 820.25 says, “(b) Training. Each manufacturer shall establish procedures for identifying training needs and ensure that all personnel are trained to adequately perform their assigned responsibilities. Training shall be documented. (1) As part of their training, personnel shall be made aware of device defects which may occur from the improper performance of their specific jobs. (2) Personnel who perform verification and validation activities shall be made aware of defects and errors that may be encountered as part of their job functions.” Additionally, Medical Device cGMP Subpart B—Quality System Requirements, 820.20, in part, asserts, “(b) Organization. Each manufacturer shall establish and maintain an adequate organizational structure to ensure that devices are designed and produced in accordance with the requirements of this part. (2) Resources. Each manufacturer shall provide adequate resources, including the assignment of trained personnel, for management, performance of work, and assessment activities, including internal quality audits, to meet the requirements of this part.”
Personnel responsibilities under the pharmaceutical cGMP requirements 211.28 specify, “(a) There shall be a quality control unit that shall have the responsibility and authority to approve or reject all components, drug product containers, closures, in-process materials, packaging material, labeling, and drug products, and the authority to review production records to assure that no errors have occurred or, if errors have occurred, that they have been fully investigated. The quality control unit shall be responsible for approving or rejecting drug products manufactured, processed, packed, or held under contract by another company. (b) Adequate laboratory facilities for the testing and approval (or rejection) of components, drug product containers, closures, packaging materials, in-process materials, and drug products shall be available to the quality control unit. (c) The quality control unit shall have the responsibility for approving or rejecting all procedures or specifications impacting on the identity, strength, quality, and purity of the drug product. (d) The responsibilities and procedures applicable to the quality control unit shall be in writing; such written procedures shall be followed.”
The Medical Device cGMP Subpart B—Quality System Requirements 820.20 address responsibilities as follows, “(b) Organization. Each manufacturer shall establish and maintain an adequate organizational structure to ensure that devices are designed and produced in accordance with the requirements of this part. (1) Responsibility and authority. Each manufacturer shall establish the appropriate responsibility, authority, and interrelation of all personnel who manage, perform, and assess work affecting quality, and provide the independence and authority necessary to perform these tasks.”
820.20 also contains the following requirement for a management representative, “(b)(3) Management representative. Management with executive responsibility shall appoint, and document such appointment of, a member of management who, irrespective of other responsibilities, shall have established authority over and responsibility for: (i) Ensuring that quality system requirements are effectively established and effectively maintained in accordance with this part; and (ii) Reporting on the performance of the quality system to management with executive responsibility for review. (c) Management review. Management with executive responsibility shall review the suitability and effectiveness of the quality system at defined intervals and with sufficient frequency according to established procedures to ensure the quality system satisfies the requirements of this part and the manufacturer’s established quality policy and objectives. The dates and results of quality system reviews shall be documented.”
Once again, the language is different, but the results are substantially equivalent. The most notable difference is only the medical device cGMP has a requirement for a management representative, (the pharmaceutical cGMP regulations lack such a mandate). In practice, however, pharmaceutical companies usually have a designated management representative.
Pharmaceutical cGMP 211.34 states, “Consultants advising on the manufacture, processing, packing, or holding of drug products shall have sufficient education, training, and experience, or any combination thereof, to advise on the subject for which they are retained. Records shall be maintained stating the name, address, and qualifications of any consultants and the type of service they provide.” Medical Device cGMP Subpart E—Purchasing Controls 820.50 states in part, “Each manufacturer shall establish and maintain procedures to ensure that all purchased or otherwise received product and services conform to specified requirements. (a) Evaluation of suppliers, contractors, and consultants. Each manufacturer shall establish and maintain the requirements, including quality requirements, that must be met by suppliers, contractors, and consultants. Each manufacturer shall: (1) Evaluate and select potential suppliers, contractors, and consultants on the basis of their ability to meet specified requirements, including quality requirements. The evaluation shall be documented. (2) Define the type and extent of control to be exercised over the product, services, suppliers, contractors, and consultants, based on the evaluation results. (3) Establish and maintain records of acceptable suppliers, contractors, and consultants.
Both the pharmaceutical and medical device cGMP requirements adequately address qualification of consultants. The medical device cGMP requirements clearly identify consultants as suppliers, while the pharmaceutical cGMP regulations classify consultants as personnel.
In summary, the pharmaceutical and medical device cGMP requirements for organization and personnel are substantially equivalent. The specific medical device cGMP requirements for a management representative and the internal quality audits mandate are effective. In practice, drug companies have a designated management representative who performs internal quality audits.
Further Reading
Review any of the other sections of this series by clicking on the headline.
Current Good Manufacturing Practices: Pharma vs. Device
Comparing cGMP Pharma vs. Device: Subpart A – General Provisions
Comparing cGMP Pharma vs. Device: Subpart A—General Provisions (Part II)
Comparing cGMP Pharma vs. Device: Subpart C—Buildings and Facilities and Subpart D—Equipment
Comparing cGMP Pharma vs. Device: Subpart E—Control of Components, Containers and Closures
James A. “Jim” Dunning’s consulting career began in 2001. He has provided quality and regulatory consulting services for various companies ranging from Fortune 500 medical device firms to startups. Dunning’s passion, however, lies with startups and small companies, especially those in regulatory distress. He has amassed significant experience in preparing 510(k) applications, developing complete Quality Management Systems, providing Quality System Training, and advising on quality, business, and leadership issues. Dunning is a senior member of the American Society for Quality (ASQ) and a member of the Regulatory Affairs Professional Society (RAPS). He can be reached at jdunning@qpcservices.com.
There are differences, too: Research and development, brand marketing, product design (mechanical vs. chemical), and technological innovation, to name just a few. Perhaps the most striking disparity, however, exists in FDA regulations, though I contend there are more similarities between the two industries than meets the eye. Hence my motivation for this ongoing series comparing parts of the drug and device industries’ Current Good Manufacturing Practices (cGMP) requirements.
This month’s column will focus on ways 21 CFR Part 820 Quality System Regulation, also known as the cGMP regulation for medical devices, aligns with the cGMP requirements of 21 CFR Part 211 Subpart B—Organization and Personnel. The two most recent articles dealt with Subpart A—General Provisions. As was the case in previous columns, the term “pharmaceutical(s)” is used rather than “drug(s)” unless the term is part of an excerpt from the cGMP regulation. Scope and definition are the same as well.
An overview of the scope of this article is provided in Table 1 (below).
From a high-level overview, the table shows that Medical Device cGMP Subpart B—Quality System Regulation aligns relatively well with Pharmaceutical cGMP Subpart B Organization and Personnel, with the exception of 211.34 Consultants. The medical device cGMP addresses consultants in Subpart E—Purchasing controls. Additionally, the pharmaceutical cGMP does not address quality audits, whereas the Medical Device cGMP Subpart B—Quality System Regulation, 820.22 Quality audit, specifically refers to quality audits. In practice, quality audits are a key Quality Control Unit responsibility, and the expectations for quality auditing are contained in U.S. Food and Drug Administration (FDA) guidance documents.
Pharmaceutical cGMP Requirements, Subpart B—Organization and Personnel, 211.22 (a) declares, “There shall be a quality control unit that shall have the responsibility and authority to approve or reject all components, drug product containers, closures, in-process materials, packaging material, labeling, and drug products, and the authority to review production records to assure that no errors have occurred or, if errors have occurred, that they have been fully investigated. The quality control unit shall be responsible for approving or rejecting drug products manufactured, processed, packed, or held under contract by another company.”
Medical Device cGMP Requirements, Subpart B Quality System Regulations, 820.20 in part, states the following:
“(b) Organization. Each manufacturer shall establish and maintain an adequate organizational structure to ensure that devices are designed and produced in accordance with the requirements of this part. (1) Responsibility and authority. Each manufacturer shall establish the appropriate responsibility, authority, and interrelation of all personnel who manage, perform, and assess work affecting quality, and provide the independence and authority necessary to perform these tasks. (2) Resources. Each manufacturer shall provide adequate resources, including the assignment of trained personnel, for management, performance of work, and assessment activities, including internal quality audits, to meet the requirements of this part. (3) Management representative. Management with executive responsibility shall appoint, and document such appointment of, a member of management who, irrespective of other responsibilities, shall have established authority over and responsibility for: (i) Ensuring that quality system requirements are effectively established and effectively maintained in accordance with this part; and (ii) Reporting on the performance of the quality system to management with executive responsibility for review.”
The approach is different, but the results are similar. I believe the pharmaceutical cGMP mandates more clearly establish the Quality Control Unit requirements, and the directives are more specific, with the exception of the management representative role. In practice, pharmaceutical companies have management representatives.
Personnel qualifications and personnel responsibilities are equally addressed in both the pharmaceutical and medical device cGMP requirements.
For personnel qualifications, Pharmaceutical cGMP 211.25 states in part, “(b) Each person responsible for supervising the manufacture, processing, packing, or holding of a drug product shall have the education, training, and experience, or any combination thereof, to perform assigned functions in such a manner as to provide assurance that the drug product has the safety, identity, strength, quality, and purity that it purports or is represented to possess.” The corresponding prerequisites under Medical Device cGMP 820.25 says, “(b) Training. Each manufacturer shall establish procedures for identifying training needs and ensure that all personnel are trained to adequately perform their assigned responsibilities. Training shall be documented. (1) As part of their training, personnel shall be made aware of device defects which may occur from the improper performance of their specific jobs. (2) Personnel who perform verification and validation activities shall be made aware of defects and errors that may be encountered as part of their job functions.” Additionally, Medical Device cGMP Subpart B—Quality System Requirements, 820.20, in part, asserts, “(b) Organization. Each manufacturer shall establish and maintain an adequate organizational structure to ensure that devices are designed and produced in accordance with the requirements of this part. (2) Resources. Each manufacturer shall provide adequate resources, including the assignment of trained personnel, for management, performance of work, and assessment activities, including internal quality audits, to meet the requirements of this part.”
Personnel responsibilities under the pharmaceutical cGMP requirements 211.28 specify, “(a) There shall be a quality control unit that shall have the responsibility and authority to approve or reject all components, drug product containers, closures, in-process materials, packaging material, labeling, and drug products, and the authority to review production records to assure that no errors have occurred or, if errors have occurred, that they have been fully investigated. The quality control unit shall be responsible for approving or rejecting drug products manufactured, processed, packed, or held under contract by another company. (b) Adequate laboratory facilities for the testing and approval (or rejection) of components, drug product containers, closures, packaging materials, in-process materials, and drug products shall be available to the quality control unit. (c) The quality control unit shall have the responsibility for approving or rejecting all procedures or specifications impacting on the identity, strength, quality, and purity of the drug product. (d) The responsibilities and procedures applicable to the quality control unit shall be in writing; such written procedures shall be followed.”
The Medical Device cGMP Subpart B—Quality System Requirements 820.20 address responsibilities as follows, “(b) Organization. Each manufacturer shall establish and maintain an adequate organizational structure to ensure that devices are designed and produced in accordance with the requirements of this part. (1) Responsibility and authority. Each manufacturer shall establish the appropriate responsibility, authority, and interrelation of all personnel who manage, perform, and assess work affecting quality, and provide the independence and authority necessary to perform these tasks.”
820.20 also contains the following requirement for a management representative, “(b)(3) Management representative. Management with executive responsibility shall appoint, and document such appointment of, a member of management who, irrespective of other responsibilities, shall have established authority over and responsibility for: (i) Ensuring that quality system requirements are effectively established and effectively maintained in accordance with this part; and (ii) Reporting on the performance of the quality system to management with executive responsibility for review. (c) Management review. Management with executive responsibility shall review the suitability and effectiveness of the quality system at defined intervals and with sufficient frequency according to established procedures to ensure the quality system satisfies the requirements of this part and the manufacturer’s established quality policy and objectives. The dates and results of quality system reviews shall be documented.”
Once again, the language is different, but the results are substantially equivalent. The most notable difference is only the medical device cGMP has a requirement for a management representative, (the pharmaceutical cGMP regulations lack such a mandate). In practice, however, pharmaceutical companies usually have a designated management representative.
Pharmaceutical cGMP 211.34 states, “Consultants advising on the manufacture, processing, packing, or holding of drug products shall have sufficient education, training, and experience, or any combination thereof, to advise on the subject for which they are retained. Records shall be maintained stating the name, address, and qualifications of any consultants and the type of service they provide.” Medical Device cGMP Subpart E—Purchasing Controls 820.50 states in part, “Each manufacturer shall establish and maintain procedures to ensure that all purchased or otherwise received product and services conform to specified requirements. (a) Evaluation of suppliers, contractors, and consultants. Each manufacturer shall establish and maintain the requirements, including quality requirements, that must be met by suppliers, contractors, and consultants. Each manufacturer shall: (1) Evaluate and select potential suppliers, contractors, and consultants on the basis of their ability to meet specified requirements, including quality requirements. The evaluation shall be documented. (2) Define the type and extent of control to be exercised over the product, services, suppliers, contractors, and consultants, based on the evaluation results. (3) Establish and maintain records of acceptable suppliers, contractors, and consultants.
Both the pharmaceutical and medical device cGMP requirements adequately address qualification of consultants. The medical device cGMP requirements clearly identify consultants as suppliers, while the pharmaceutical cGMP regulations classify consultants as personnel.
In summary, the pharmaceutical and medical device cGMP requirements for organization and personnel are substantially equivalent. The specific medical device cGMP requirements for a management representative and the internal quality audits mandate are effective. In practice, drug companies have a designated management representative who performs internal quality audits.
Further Reading
Review any of the other sections of this series by clicking on the headline.
Current Good Manufacturing Practices: Pharma vs. Device
Comparing cGMP Pharma vs. Device: Subpart A – General Provisions
Comparing cGMP Pharma vs. Device: Subpart A—General Provisions (Part II)
Comparing cGMP Pharma vs. Device: Subpart C—Buildings and Facilities and Subpart D—Equipment
Comparing cGMP Pharma vs. Device: Subpart E—Control of Components, Containers and Closures
James A. “Jim” Dunning’s consulting career began in 2001. He has provided quality and regulatory consulting services for various companies ranging from Fortune 500 medical device firms to startups. Dunning’s passion, however, lies with startups and small companies, especially those in regulatory distress. He has amassed significant experience in preparing 510(k) applications, developing complete Quality Management Systems, providing Quality System Training, and advising on quality, business, and leadership issues. Dunning is a senior member of the American Society for Quality (ASQ) and a member of the Regulatory Affairs Professional Society (RAPS). He can be reached at jdunning@qpcservices.com.