
By Sean Fenske, Editor-in-Chief
With a new product design comes an array of challenges as well as questions. A designer needs to address all of them within the device ideation. Determining the interface, identifying the primary user, considering human factors, choice of materials, and many more all need to be considered and decided.
Another extremely important aspect is knowing what device classification the product will fall under. As such, the design may have additional considerations that need to be addressed and quality gates it will pass through. The higher the classification, the more challenges tied to the design. As such, best practices must be employed to avoid problems further down the line that could cost time and money in getting the product to market (or even through the appropriate regulatory agency).
Fortunately, Peter van Beek, Business Development Manager of Medical for maxon, took time to respond to questions around risk, device classification, and design. He offers insights on the relationship between class and design while also providing important considerations engineers should keep top of mind.
Sean Fenske: When designing a motor assembly into a medical device, what are the first critical considerations that must be determined before design progresses?
Peter van Beek: There needs to be clear understanding of the needs for the overall design between supplier and user for their specific market. This understanding would best be summarized at the start of the project with a functional specification. Most projects start with a complete exchange of all required parameters. The most critical parameters being voltage, torque, speed, diameter, and length. Additional critical considerations mainly associated with power tools and surgical robot end effectors are the need for sterilization, exposure to saline, or corrosive chemistry. For radiation emitting applications, the radiation life dose and exposure levels are needed.
Fenske: Can you explain what is meant by high-risk class for both FDA and MDR and how these are applied?
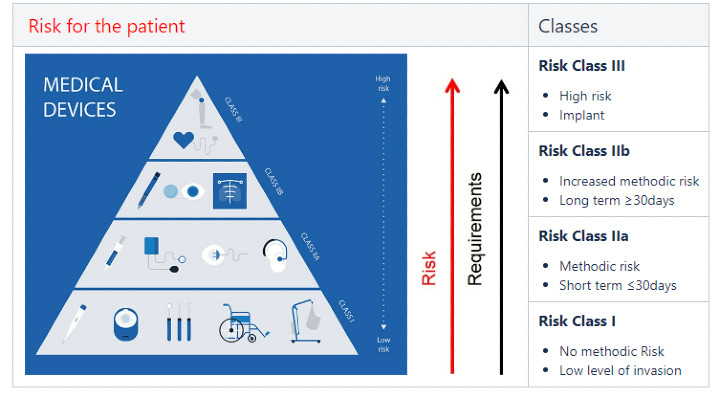
van Beek: Both the FDA and MDR (medical device regulation; EU-based) utilize a number system for device Class, consisting of I (low risk), II (medium to increased risk), and III (high risk). The MDR differs only from the FDA numbering by employing two designations for II (IIA and IIB). The higher the number, the greater the risk level. Typical examples of Class I devices would be a wheelchair or thermometer; Class IIA examples (short term use ≤30 days) are contact lenses or pregnancy tests; Class IIB examples (long term use ≥30 days, increased risk) insulin pumps or surgical robots; and Class III examples (high risk) include implanted devices such as a heart pump or pacemaker.
Fenske: With regard to the EU MDR, what new device regulations will affect the design process and do you have tips for engineers to address them?
van Beek: The European MDR will replace the EU’s current Medical Device Directive and the EU’s directive on implantable devices. The regulation applies to all medical device manufacturers who intend to sell their medical devices in the European market (i.e., EU nations). The new regulation dictates a rigorous post product launch oversight by the regulating body. A design will need to be robust and, over time, continue to be capable of providing the benefit claim and safety like a new device. The clinical evidence requirements for Class III devices, including implantable devices, are increasing. Class IIA and IIB devices will be required to undergo a systematic clinical evaluation process. Finally, there will be no grandfathering provision for existing tools already on the market.
Fenske: When designing high-risk class devices, how is a risk management strategy used to help ensure success?
van Beek: maxon employs a milestones approach to projects starting with a QAA (Quality Assurance Agreement). FMEAs (failure modes and effects analysis) are performed on both the design and processes to completely understand all associated risks and determine strategies to nullify them. The capability of the design will be confirmed—for example, motor assembly lifetime testing in the actual end-product. Typically for the medical industry, maxon performs both OQ (operational qualification) and PQ (performance qualification) validations. After a qualified process (via validation methods) has been established, the first parts are built and analyzed to create a FAI (first article inspection).

maxon offers a complete family of motor and motion control solutions to meet the needs of every medical device manufacturer developing a new healthcare product.
Fenske: When considering a device’s design development along with the quality requirements, what are your suggestions for best practices?
van Beek: Start the project with a FRS (functional requirement specification) between your primary suppliers to define development targets. A Quality Assurance Agreement should be completed prior to (or at the latest) the design freeze. This aligns the two companies’ quality expectations and allows maxon to start both design and process FMEA analysis and, later, validation planning. This also provides the customer with an indication of costs associated for the validation, which can be substantial. Coordination early results in smooth flow through these required gates and associated costs.
Fenske: How do validation, verification, test, and documentation factor into the medical device development “recipe.” At what point do these aspects get addressed or determined?
van Beek: maxon again likes to start projects with a Specification & Quality Assurance Agreement which sets the stage for later validation requirements. Process validation can start early in a project, but it really makes no sense to do this until the customer has completed design validation and frozen the design. Once the design is locked, complete validation can start on production lines, fixtures/toolings, test equipment, and even operators. The validation steps confirm the process of building is sound and capable. Once a validated production line is complete, maxon is ready to produce qualified parts.
Fenske: Do you have any additional comments you’d like to share based on any of the topics we discussed or something you’d like to tell medical device manufacturers?
van Beek: The majority of device developments start out design heavy, but with little consideration of required quality gates and eventual validations in mind. A successful medical design must walk hand-in-hand with quality from start to finish. Again, don’t leave your quality and validation until the end of the project.
Click here to find out more about maxon >>>>>
With a new product design comes an array of challenges as well as questions. A designer needs to address all of them within the device ideation. Determining the interface, identifying the primary user, considering human factors, choice of materials, and many more all need to be considered and decided.
Another extremely important aspect is knowing what device classification the product will fall under. As such, the design may have additional considerations that need to be addressed and quality gates it will pass through. The higher the classification, the more challenges tied to the design. As such, best practices must be employed to avoid problems further down the line that could cost time and money in getting the product to market (or even through the appropriate regulatory agency).
Fortunately, Peter van Beek, Business Development Manager of Medical for maxon, took time to respond to questions around risk, device classification, and design. He offers insights on the relationship between class and design while also providing important considerations engineers should keep top of mind.
Sean Fenske: When designing a motor assembly into a medical device, what are the first critical considerations that must be determined before design progresses?
Peter van Beek: There needs to be clear understanding of the needs for the overall design between supplier and user for their specific market. This understanding would best be summarized at the start of the project with a functional specification. Most projects start with a complete exchange of all required parameters. The most critical parameters being voltage, torque, speed, diameter, and length. Additional critical considerations mainly associated with power tools and surgical robot end effectors are the need for sterilization, exposure to saline, or corrosive chemistry. For radiation emitting applications, the radiation life dose and exposure levels are needed.
Fenske: Can you explain what is meant by high-risk class for both FDA and MDR and how these are applied?
van Beek: Both the FDA and MDR (medical device regulation; EU-based) utilize a number system for device Class, consisting of I (low risk), II (medium to increased risk), and III (high risk). The MDR differs only from the FDA numbering by employing two designations for II (IIA and IIB). The higher the number, the greater the risk level. Typical examples of Class I devices would be a wheelchair or thermometer; Class IIA examples (short term use ≤30 days) are contact lenses or pregnancy tests; Class IIB examples (long term use ≥30 days, increased risk) insulin pumps or surgical robots; and Class III examples (high risk) include implanted devices such as a heart pump or pacemaker.
Fenske: With regard to the EU MDR, what new device regulations will affect the design process and do you have tips for engineers to address them?
van Beek: The European MDR will replace the EU’s current Medical Device Directive and the EU’s directive on implantable devices. The regulation applies to all medical device manufacturers who intend to sell their medical devices in the European market (i.e., EU nations). The new regulation dictates a rigorous post product launch oversight by the regulating body. A design will need to be robust and, over time, continue to be capable of providing the benefit claim and safety like a new device. The clinical evidence requirements for Class III devices, including implantable devices, are increasing. Class IIA and IIB devices will be required to undergo a systematic clinical evaluation process. Finally, there will be no grandfathering provision for existing tools already on the market.
Fenske: When designing high-risk class devices, how is a risk management strategy used to help ensure success?
van Beek: maxon employs a milestones approach to projects starting with a QAA (Quality Assurance Agreement). FMEAs (failure modes and effects analysis) are performed on both the design and processes to completely understand all associated risks and determine strategies to nullify them. The capability of the design will be confirmed—for example, motor assembly lifetime testing in the actual end-product. Typically for the medical industry, maxon performs both OQ (operational qualification) and PQ (performance qualification) validations. After a qualified process (via validation methods) has been established, the first parts are built and analyzed to create a FAI (first article inspection).
maxon offers a complete family of motor and motion control solutions to meet the needs of every medical device manufacturer developing a new healthcare product.
Fenske: When considering a device’s design development along with the quality requirements, what are your suggestions for best practices?
van Beek: Start the project with a FRS (functional requirement specification) between your primary suppliers to define development targets. A Quality Assurance Agreement should be completed prior to (or at the latest) the design freeze. This aligns the two companies’ quality expectations and allows maxon to start both design and process FMEA analysis and, later, validation planning. This also provides the customer with an indication of costs associated for the validation, which can be substantial. Coordination early results in smooth flow through these required gates and associated costs.
Fenske: How do validation, verification, test, and documentation factor into the medical device development “recipe.” At what point do these aspects get addressed or determined?
van Beek: maxon again likes to start projects with a Specification & Quality Assurance Agreement which sets the stage for later validation requirements. Process validation can start early in a project, but it really makes no sense to do this until the customer has completed design validation and frozen the design. Once the design is locked, complete validation can start on production lines, fixtures/toolings, test equipment, and even operators. The validation steps confirm the process of building is sound and capable. Once a validated production line is complete, maxon is ready to produce qualified parts.
Fenske: Do you have any additional comments you’d like to share based on any of the topics we discussed or something you’d like to tell medical device manufacturers?
van Beek: The majority of device developments start out design heavy, but with little consideration of required quality gates and eventual validations in mind. A successful medical design must walk hand-in-hand with quality from start to finish. Again, don’t leave your quality and validation until the end of the project.
Click here to find out more about maxon >>>>>

