Seth J. Goldenberg, Ph.D., Vice President, Vault Medical Device & Diagnostics, Veeva Systems07.30.19
Medical device products are rapidly evolving to become more personalized and connected, leveraging artificial intelligence (AI), robotics, and mobile technology.
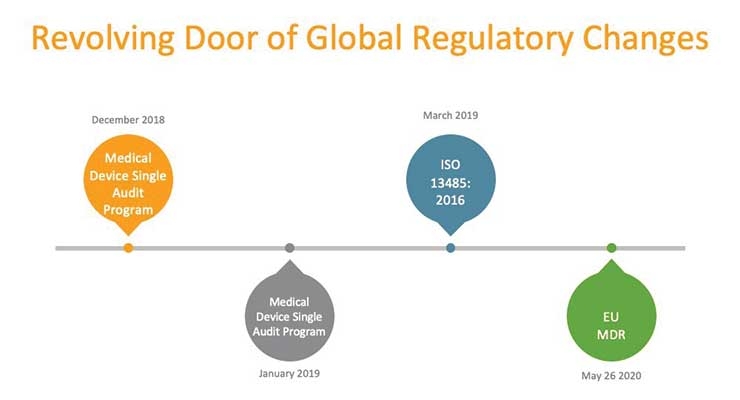
Against this backdrop of continual innovation, medical device manufacturers face unique challenges to comply with significant regulatory changes in Europe and the United States—some that could support new product development, while others could hinder it.
These challenges also offer a significant opportunity to streamline business processes and data management for improved compliance, stronger collaboration, and greater efficiency.
Modernizing Product Development
Evolving compliance rules such as the new Medical Device Regulation (MDR) in Europe include sweeping changes regarding how manufacturers and distributors demonstrate the safety and efficacy of products. Companies will be required to show clinical evidence for all products as well as maintain systems and documents to remain compliant throughout the device lifecycle.
One way companies are responding is by adopting modern technology solutions for improved nimbleness and flexibility. Roche Diagnostics, for one, found that unified cloud applications can help maintain compliance and better facilitate collaboration across geographies. An evolving regulatory environment means it was no longer feasible to have disparate content without central access, the company said.
“When our regulators and legal team started looking at a lot of these things, it was through the pharma lens of the business, which is larger, not medical device/diagnostic,” said Ayaz Malik, head of diagnostic digital communications at Roche Diagnostics. “We had to explain we are regulated differently, and we don’t have the same burden as pharma.”
Whether regulatory authorities are tightening rules around medical devices or responding to market innovation, manufacturers must be prepared to demonstrate their products are safe and effective across the total lifecycle using a wider array of data sources.
Five strategies can help meet these compliance challenges while streamlining business processes and creating efficiency.
1. Gain a Complete View of Data
With the scale and complexity of data expanding, medical device companies are driving to assemble and analyze data faster. Much of this data is spread across information silos. As a result, device companies are increasingly looking to modern clinical data management systems, including an electronic data capture (EDC), that collect and clean this clinical data, all in one system.
Nearly 60 percent of medical device manufacturers say clinical data management rules are the most challenging component of Europe’s new MDR. An equal percentage of companies say they don’t have a strategy for compliance.1 Having a strategy that includes a complete view of data can help with preparation and ongoing compliance.
2. Establish a Single Source of Truth for Greater Insights
During a clinical trial, many disparate documents are gathered together into a trial master file (TMF) and submitted to regulators. In an eTMF, processes and documentation move from paper to electronic. An active eTMF operating model manages all processes and documents in real time, as they are executed, to maintain a constant state of inspection-readiness.
Documentation is a particularly important aspect of MDR, requiring that documents on a variety of business processes, including clinical trials, finances, outsourcing strategies, and even organizational design, are easily accessible to health authorities. Documentation, clinical evaluation, and traceability of devices require close alignment of fields that are often siloed today. However, an active eTMF model brings all documents together in one easily accessible system.
Improved visibility into trial documentation and processes strengthens collaboration among clinical teams and other partners. One top 20 medical device company reported $1.2 million in annual cost reduction within two years of adopting an active eTMF model.2
3. Elevate Quality Processes
The U.S. Food and Drug Administration (FDA), and Europe under MDR, are demanding more from manufacturers throughout their quality control processes, including more audit trails across the product lifecycle.
Seeking to strike a balance, the FDA’s Center for Devices and Radiological Health (CDRH) in May operationalized a new “super office” responsible for post-market medical device and radiological product surveillance and communication. The Office of Product Evaluation and Quality (OPEQ) aims to harmonize pre- and post-market surveillance to more quickly address and resolve emerging safety issues and communicate them to relevant stakeholders.3
In forming OPEQ, the FDA noted that many aspects of medical device oversight date back to the 1970s, and are no longer adequate to ensure that complex, 21st century devices are safe and effective and responsive to a rapidly innovating market.4
Unifying the quality environment and bringing teams, data, and documents together on a single modern platform creates greater efficiencies, control, and collaboration. This also eliminates silos to speed the approval process across regulatory, production, engineering, and quality. All stakeholders can be actively involved in the process for fewer re-approvals and re-reviews.
4. Integrate Medical Content Management
Medical affairs teams struggle to access consistent and accurate scientific information. A single and secure solution accessible to all stakeholders facilitates the authoring, review, approval, distribution, and access of medical content globally.
A centralized source where all versions are updated at once and across channels also reduces compliance risk and provides consistency. Seamless integration with authoring programs allows for compliant and real-time collaborative authoring.
5. Unify Commercial Content and Data
Medical device companies are creating more content each year across a growing number of digital channels. Managing this surplus of content and assets efficiently can be a struggle. More than 75 percent of surveyed leaders said their organizations produce “moderate to enormous” amounts of digital content, but just 13 percent said they think the content is managed well.5
Systems that bring together disparate commercial content allow medical device companies to improve compliance by updating assets simultaneously across channels and geographies. Combining digital asset management with medical, legal, and regulatory review reduces risk and increases content reuse.
Medical device regulations are constantly evolving. Approaching new regulatory challenges as opportunities to streamline systems and processes can add efficiencies that improve compliance while also supporting collaboration and innovation in the years to come.
References
Seth J. Goldenberg is responsible for Veeva Vault strategy in the medical device and diagnostic industry, including customer engagement, market adoption, and product development. Goldenberg has nearly 20 years of experience supporting medical device and diagnostic companies to navigate complex regulations and improve market access. Before joining Veeva, Goldenberg was director of product marketing at North American Science Associates (NAMSA), where he supported medical device companies from inception through commercialization and post-market activities. Goldenberg also has experience supporting life science companies to achieve their commercial goals from his time as a consultant. Outside of Veeva, Goldenberg plays an active role in the industry as member of the Regulatory Affairs Professionals Society (RAPS) and as the “entrepreneur in residence” at the Philadelphia Pediatric Device Consortium. He holds a doctorate in pharmacology from the University of Washington and a master’s degree in biomedical engineering from Drexel University. He can be reached at seth.goldenberg@veeva.com.
Against this backdrop of continual innovation, medical device manufacturers face unique challenges to comply with significant regulatory changes in Europe and the United States—some that could support new product development, while others could hinder it.
These challenges also offer a significant opportunity to streamline business processes and data management for improved compliance, stronger collaboration, and greater efficiency.
Modernizing Product Development
Evolving compliance rules such as the new Medical Device Regulation (MDR) in Europe include sweeping changes regarding how manufacturers and distributors demonstrate the safety and efficacy of products. Companies will be required to show clinical evidence for all products as well as maintain systems and documents to remain compliant throughout the device lifecycle.
One way companies are responding is by adopting modern technology solutions for improved nimbleness and flexibility. Roche Diagnostics, for one, found that unified cloud applications can help maintain compliance and better facilitate collaboration across geographies. An evolving regulatory environment means it was no longer feasible to have disparate content without central access, the company said.
“When our regulators and legal team started looking at a lot of these things, it was through the pharma lens of the business, which is larger, not medical device/diagnostic,” said Ayaz Malik, head of diagnostic digital communications at Roche Diagnostics. “We had to explain we are regulated differently, and we don’t have the same burden as pharma.”
Whether regulatory authorities are tightening rules around medical devices or responding to market innovation, manufacturers must be prepared to demonstrate their products are safe and effective across the total lifecycle using a wider array of data sources.
Five strategies can help meet these compliance challenges while streamlining business processes and creating efficiency.
1. Gain a Complete View of Data
With the scale and complexity of data expanding, medical device companies are driving to assemble and analyze data faster. Much of this data is spread across information silos. As a result, device companies are increasingly looking to modern clinical data management systems, including an electronic data capture (EDC), that collect and clean this clinical data, all in one system.
Nearly 60 percent of medical device manufacturers say clinical data management rules are the most challenging component of Europe’s new MDR. An equal percentage of companies say they don’t have a strategy for compliance.1 Having a strategy that includes a complete view of data can help with preparation and ongoing compliance.
2. Establish a Single Source of Truth for Greater Insights
During a clinical trial, many disparate documents are gathered together into a trial master file (TMF) and submitted to regulators. In an eTMF, processes and documentation move from paper to electronic. An active eTMF operating model manages all processes and documents in real time, as they are executed, to maintain a constant state of inspection-readiness.
Documentation is a particularly important aspect of MDR, requiring that documents on a variety of business processes, including clinical trials, finances, outsourcing strategies, and even organizational design, are easily accessible to health authorities. Documentation, clinical evaluation, and traceability of devices require close alignment of fields that are often siloed today. However, an active eTMF model brings all documents together in one easily accessible system.
Improved visibility into trial documentation and processes strengthens collaboration among clinical teams and other partners. One top 20 medical device company reported $1.2 million in annual cost reduction within two years of adopting an active eTMF model.2
3. Elevate Quality Processes
The U.S. Food and Drug Administration (FDA), and Europe under MDR, are demanding more from manufacturers throughout their quality control processes, including more audit trails across the product lifecycle.
Seeking to strike a balance, the FDA’s Center for Devices and Radiological Health (CDRH) in May operationalized a new “super office” responsible for post-market medical device and radiological product surveillance and communication. The Office of Product Evaluation and Quality (OPEQ) aims to harmonize pre- and post-market surveillance to more quickly address and resolve emerging safety issues and communicate them to relevant stakeholders.3
In forming OPEQ, the FDA noted that many aspects of medical device oversight date back to the 1970s, and are no longer adequate to ensure that complex, 21st century devices are safe and effective and responsive to a rapidly innovating market.4
Unifying the quality environment and bringing teams, data, and documents together on a single modern platform creates greater efficiencies, control, and collaboration. This also eliminates silos to speed the approval process across regulatory, production, engineering, and quality. All stakeholders can be actively involved in the process for fewer re-approvals and re-reviews.
4. Integrate Medical Content Management
Medical affairs teams struggle to access consistent and accurate scientific information. A single and secure solution accessible to all stakeholders facilitates the authoring, review, approval, distribution, and access of medical content globally.
A centralized source where all versions are updated at once and across channels also reduces compliance risk and provides consistency. Seamless integration with authoring programs allows for compliant and real-time collaborative authoring.
5. Unify Commercial Content and Data
Medical device companies are creating more content each year across a growing number of digital channels. Managing this surplus of content and assets efficiently can be a struggle. More than 75 percent of surveyed leaders said their organizations produce “moderate to enormous” amounts of digital content, but just 13 percent said they think the content is managed well.5
Systems that bring together disparate commercial content allow medical device companies to improve compliance by updating assets simultaneously across channels and geographies. Combining digital asset management with medical, legal, and regulatory review reduces risk and increases content reuse.
Medical device regulations are constantly evolving. Approaching new regulatory challenges as opportunities to streamline systems and processes can add efficiencies that improve compliance while also supporting collaboration and innovation in the years to come.
References
- Emergo Blog (October 10, 2018), “Medical Device Companies Underprepared for MDR,” by Stewart Eisenhart.
- Veeva Systems
- FDA Office of Product Evaluation and Quality http://bit.ly/mpo190790
- Implementing a Team-Based Approach to Medical Device and Radiological Product Evaluation and Safety http://bit.ly/mpo190791
- Accenture, “The State of Content Survey for Life Sciences,” December 2016.
Seth J. Goldenberg is responsible for Veeva Vault strategy in the medical device and diagnostic industry, including customer engagement, market adoption, and product development. Goldenberg has nearly 20 years of experience supporting medical device and diagnostic companies to navigate complex regulations and improve market access. Before joining Veeva, Goldenberg was director of product marketing at North American Science Associates (NAMSA), where he supported medical device companies from inception through commercialization and post-market activities. Goldenberg also has experience supporting life science companies to achieve their commercial goals from his time as a consultant. Outside of Veeva, Goldenberg plays an active role in the industry as member of the Regulatory Affairs Professionals Society (RAPS) and as the “entrepreneur in residence” at the Philadelphia Pediatric Device Consortium. He holds a doctorate in pharmacology from the University of Washington and a master’s degree in biomedical engineering from Drexel University. He can be reached at seth.goldenberg@veeva.com.