Matt Therrien & The Medical OEM Consortium07.26.17
Much has been written and said regarding the “what and how-to” as it relates to process development and moving a mold between machines for the medical device industry. Two years ago, a molder asked if RJG had documentation to help prove to its medical original equipment manufacturer (OEM) customer that if they moved a mold from one facility to another, they wouldn’t have to revalidate the process or, at a minimum, they could offer a reduced verification run. After some due diligence, a model started to be formed, with the ultimate goal for it to be universally accepted by the medical device industry.
The economics of adopting this approach could potentially not only save tens to hundreds of thousands of dollars for each move (depending upon the number of molds), but the speed-to-market advantages and operations flexibility would be simply invaluable.
In the world of medical device manufacturing, the control over changes in manufacturing are restrictive, forcing manufacturers to prove that changes introduced to the process will not impact the design or performance of the device. While necessary to ensure patient and consumer safety, in the case of injection molding, these requirements are costly and create a significant strain on resources—both at the OEM and supplier level.
The idea for the new approach for moving molds between machines was then reviewed with individual medical device OEMs over the next year. That is also when the Consortium was proposed—to enable the OEM members to physically test the concept first hand. Out of the nine that were approached, six accepted the invitation to participate. In addition, there was one molder who maintained multiple facilities and machines in close proximity to the Consortium team, and a mold maker that could support the mold. While there is existing information on this idea, each OEM wanted to put the “proof of concept” to the test and fully document the results as a case study that could be exercised and put into practice.
The OEM Consortium Team
The team started by using the principles outlined in the MPO May 2017 article “A Team Approach to Product Development” to employ the benefits of collaboration. This was executed at a high level within the medical OEM Consortium (MOEMC); the leaders of this initiative consisted of employees from six medical OEMs—Becton Dickinson, Eli Lilly, Johnson & Johnson, Teleflex Vascular Division, and Terumo Cardiovascular Group (one chose to remain anonymous)—along with the medical molder (Nypro, a Jabil Company), and the mold maker (NyproMold), all facilitated by RJG.
Each OEM’s team was comprised of three to five senior leaders from the company, which included program management, SQE/quality management, and process engineering management, so the premise of the method could be challenged against their global needs. Each OEM was required to have representation from quality or they could not attend; the perception that “it can’t be done” is the challenge the industry has to overcome.
The premise of the Consortium was to prove that Rod Groleau’s (founder of RJG Inc.) white paper, “Location Independent PPAP Streamlined for Global Manufacturing” (presented at the University of Michigan Automotive Conference in August 2000), could also apply to the medical device industry. As a collaborative team, the MOEMC exercised a Decoupled 2 (DC2) process using available systematic molding principles and technology on four predetermined, capable machines—all of different make, model (electric/hydraulic), tonnage, barrel size, volumetric injection rates, and in different locations. The concept was to challenge the traditional philosophy of a fixed machine settings validation and move toward a more flexible “Part Process Development/Validation” for a molded part across multiple machines. The MOEMC focused on reproducing a consistent “end result” based on the transfer of a DC2 process via machine independent variables (MIV) and verification of what was happening in the cavity using technology, instead of machine inputs/settings for a defined process.
The event took into consideration the applicable regulatory requirements outlined in 21 CFR part 820 as it relates to molded plastic components typically assembled into a finished device. The end result was a well-documented case study to expose the medical device industry to the “Part Process Development/Validation” strategy as a robust alternate methodology to traditional validation concepts.
The team met in May 2016 to investigate and exercise a good/better/best approach to facilitate the validation/transfer. The four mold/machine transfer trials were completed by March 2017.
Workflow
The MOEMC not only took the time to execute the mold transfer between the four distinctly different machines, it also validated the process with data-driven results and verified it using technology. The MOEMC team established a Validation Master Plan (VMP) for the event and executed the initial full validation, followed by three separate qualification/verification trials, each with its own validation report. All information was collated in a Summary Report documenting the conclusive evidence that the MIV process was repeatable and all the dimensional inspections exceeded the targeted 1.5 Ppk and attribute data (Figure 1).
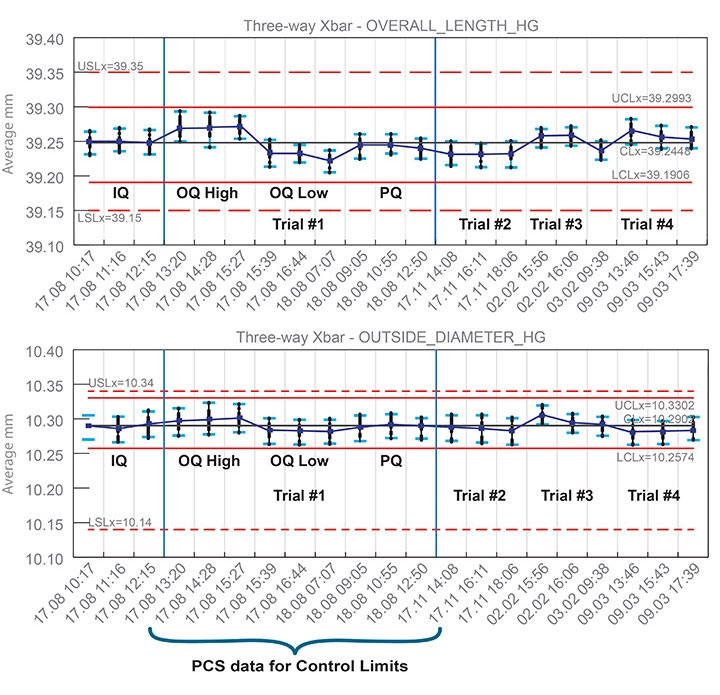
Figure 1: Four validation trials were completed. Dimensional results (beginning/middle/end of each trial) were within validation control limits. Image courtesy of RJG Inc.
Pre-trial assumptions were established with a systematic process development approach that defines a “part process” paired with a “capable” machine. The focus was on optimization of what the plastic was experiencing at the part level as a result of the applied design principles, material selection, mold design and construction, and machine capability to deliver the same end results.
The 1.4g part (drive clutch) was molded in an Acetal grade material. The mold that produced the part was a 16-cavity, direct valve gate, hot runner mold with an A-side lifter and B-side actions. Once the MIV transferred process was stable for each individual trial, sample parts were measured to confirm dimensional acceptance. Technology was then utilized to visually verify and confirm that the process was normalized between the qualification and validation machines. The machine(s) were instrumented with pressure sensors and linear transducers to monitor plastic pressure and linear displacement in the injection unit. The mold had flow meters and temperature sensors to monitor the water cooling circuits and temperature of the cavity/core steel. Additionally, each cavity was equipped with force sensors to read plastic pressure. The additional data provided by technology increased confidence that the original validated process was being replicated in Trials 2 through 4.
The technology used (eDART System) allowed the team to characterize the process and graphically visualize aspects that would never otherwise be known. Documented record (objective evidence/proof) of the interaction of the outputs/results from the “plastic’s point of view” illustrated that the part process was repeatable and consistent over time, including the full history of how and why. This provided data for a visual, data-driven, quantifiable, and tangible IQ, OQ, PQ. Electronic files retain the trail of how one arrived at the process development validation and the objective evidence to correlate the part process plastic conditions to the resultant dimensional data/report (Figure 2).
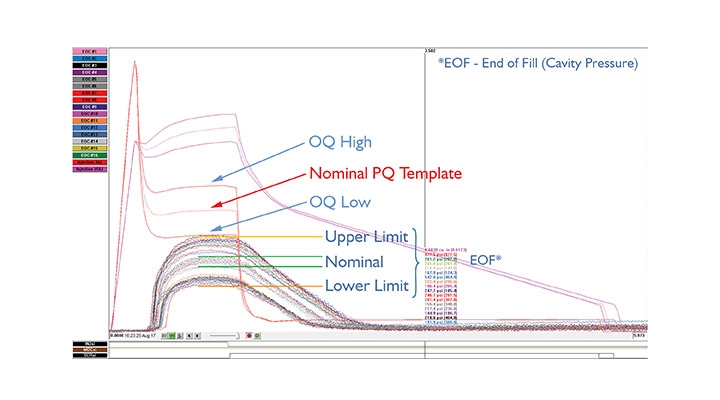
Figure 2: Validation run: Process overlay of PQ, OQ High, OQ Low. Technology allowed the team to identify the upper/lower part process control limits. Image courtesy of RJG Inc.
The Results
Based on the documented results, the team has the confidence to utilize the established method as an alternative to the traditional single machine validation approach, which will be exercised and put into practice. The first article inspection report (FAIR) dimensional data for the reports for the validation and three qualification runs was executed by 3D ProScan, a division of NyproMold, using a CT scanner that compares the actual part to the 3D model and yields highly accurate results days to weeks faster than traditional metrology methods. Process capability data was measured through standard inspection practices.
The MOEMC team effectively validated and verified that the drive clutch is capable (and ready) of running production in four different machines. There is measured and documented data, representing both dimensional and process matched MIV that is supported by the data. The method and results were documented in the Validation Master Plan (VMP), and original validation report VAL-RJG-001, qualification reports VAL-RJG-002 through -004, and summary report VAL-RJG-005.
A validation (typically defined by a part dim report) has to include the molding process itself that produced the parts. This will include the record/documentation of how it was developed—proof that it is capable of producing repeatable parts (to dim) and consistently within the established control limits. Having the full roadmap, from development to production, allows a plastic part process to be portable (dimensions are a result of the defined part process). Everything is selected for a reason and should have background data to support it. That represents a true “part process” development.
Fully documented dimensional and MIV part process-related results satisfy the criteria that is typically outlined in any Master Validation Plan (MVP). The proven results are transferable to any machine that can be verified to be capable of consistently delivering the repeatable MIV plastic variables and plastic conditions. The performance of the machine has to be documented/confirmed to be within the typical recommended target limits (i.e., acceptable shot capacity, consistent melt preparation, can hold consistent part tonnage, delivers sufficient volumetric injection rate, and is not pressure limited).
These results can be plugged into any standard validation/qualification report to satisfy the regulatory documentation requirements.
The MIV checklist becomes the guidance to making a good part. Machine adjustments (from machine to machine) can be made to stay within the acceptable tolerances (established during OQ) of the defined part process. The MIV represents the plastics variables/conditions of the “part process”: a combination of the specific input and output values that characterize the particular part/mold. If these are reproduced in a defined capable machine, the same end results will be generated (i.e., part dimensions).
The use of technology provides additional process verification that the same parts are being made. It also provides a full shot-to-shot graphical record of the process outputs that represent the “fingerprint” of the part. Using a combination of the MIV Checklist and available technology (in this case, the eDART System), one can easily and quickly reproduce the process in any machine (defined as capable). This now allows for the ability to transfer molds and increases flexibility in capacity management.
Part process development/validation truly requires the engineer to look at the process from the plastic’s point of view, not the machine’s. With a machine independent approach, the machine becomes an interchangeable component of the manufacturing cell with the mold (part) at the center. The bill of materials (BOM) of the cell is comprised of the mold, machine, material, and ancillary equipment to support it. This is typically comprised of a mold temperature control unit, hot runner controller, flow meters, material dryer, robot, EOA, etc., which have all been calibration certified to performance—a typical part of the required IQ (Figure 3).

Figure 3: Compatibility: Mold and machine match based on performance specification: BOM-capable machines are interchangeable from…the plastic’s point of view. Image courtesy of RJG Inc.
Additionally, if there is a means to monitor and verify this performance, it should be done. This is required to increase confidence levels, leading to a higher level of assurance. Now during a transfer, companies can effectively run an abbreviated PQ trial and produce with confidence. Once the formal PQ report is generated and the parts pass the VMP criteria, the parts can be approved and released into the product stream.
Using a lead measure principles approach (predictive and influenceable) versus a lag approach (checking after the fact) is comprehensive and non-restrictive. This effective principle-based systematic approach to process development and validation, based on the MIV and verification with technology, can increase the level of confidence that good parts are being introduced to the product stream.
Through this new strategy, costs and resources can be significantly reduced without impacting plastic part quality or functionality. The approach requires a fully validated part process for a mold and machine(s) with detailed development history. The approach allows reduced validation during transfer to a capable machine to improve capacity restraints within manufacturing or to a new supplier.
The ultimate paradigm shift is to focus on the plastic part process outputs (plastic conditions and component variables/attributes) versus machine set points. The focus becomes the part inside the mold, not the machine, with an objective to achieve more consistent, repeatable results. Although not required, technology provides immediate verification to the originally validated part process or immediate response to normalize plastic conditions and machine performance across multiple machines. Technology can capture shot-to-shot performance, reducing reaction time associated with part measurement and visual inspection to confirm part quality (Table).
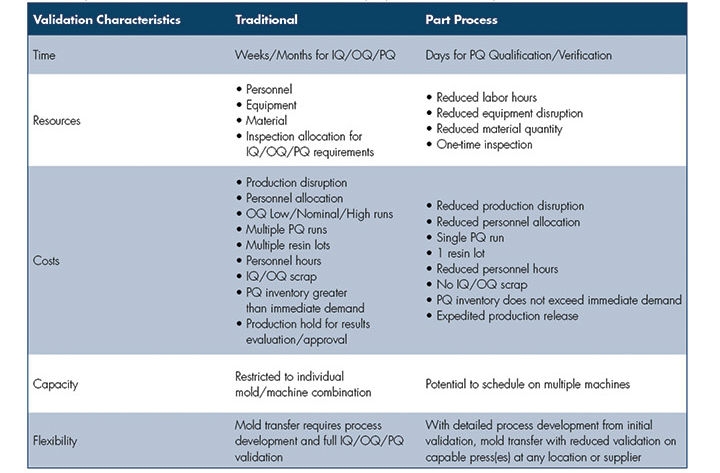
Table: A comparison of traditional mold transfer validation characteristics and
part process validation concepts.
Conclusion
Through the execution of the discovery exercise, the MOEMC has provided a broadened perspective on the acceptance of the “part process” method to allow the transfer of molds in the medical device industry as a practical alternate standard.
What was amazing to watch was that the SQE/Quality Team really dug in to learn about the molding process and its effect on the dimensional results. At the end of the Consortium, they were the most passionate about explaining the benefits of the newfound data-driven approach. Each OEM understands that they will need to execute this method (with or without technology) with one of their leading molding/converter partners to further educate internal/external teams on the principles that need to be followed.
In turn, embedding the MIV method delivers a more thorough part process development/validation that is repeatable and capable of being transferred through multiple machines. It is critical to have both the dimensional and plastic process variable (MIV) data fully documented for assurance of consistent results. Creating a case study that incorporates each OEM’s own validation protocol documentation/reporting requirements will provide a template for future program success.
The industry as a whole can now become more knowledgeable about what the levels of operational excellence (number of buildings and equipment doesn’t define this) really means when assessing a partner for manufacturing. Also, there is an increased assurance that the methods used can help reduce and mitigate risk based on data-driven decisions (using available technology) that can help drive continuous improvements and increased statistical control.
[Author’s Note: A medical device industry white paper on the subject matter is now available for download. Click here to get the white paper.]
Matt Therrien is the northeast regional manager at RJG Inc.
The economics of adopting this approach could potentially not only save tens to hundreds of thousands of dollars for each move (depending upon the number of molds), but the speed-to-market advantages and operations flexibility would be simply invaluable.
In the world of medical device manufacturing, the control over changes in manufacturing are restrictive, forcing manufacturers to prove that changes introduced to the process will not impact the design or performance of the device. While necessary to ensure patient and consumer safety, in the case of injection molding, these requirements are costly and create a significant strain on resources—both at the OEM and supplier level.
The idea for the new approach for moving molds between machines was then reviewed with individual medical device OEMs over the next year. That is also when the Consortium was proposed—to enable the OEM members to physically test the concept first hand. Out of the nine that were approached, six accepted the invitation to participate. In addition, there was one molder who maintained multiple facilities and machines in close proximity to the Consortium team, and a mold maker that could support the mold. While there is existing information on this idea, each OEM wanted to put the “proof of concept” to the test and fully document the results as a case study that could be exercised and put into practice.
The OEM Consortium Team
The team started by using the principles outlined in the MPO May 2017 article “A Team Approach to Product Development” to employ the benefits of collaboration. This was executed at a high level within the medical OEM Consortium (MOEMC); the leaders of this initiative consisted of employees from six medical OEMs—Becton Dickinson, Eli Lilly, Johnson & Johnson, Teleflex Vascular Division, and Terumo Cardiovascular Group (one chose to remain anonymous)—along with the medical molder (Nypro, a Jabil Company), and the mold maker (NyproMold), all facilitated by RJG.
Each OEM’s team was comprised of three to five senior leaders from the company, which included program management, SQE/quality management, and process engineering management, so the premise of the method could be challenged against their global needs. Each OEM was required to have representation from quality or they could not attend; the perception that “it can’t be done” is the challenge the industry has to overcome.
The premise of the Consortium was to prove that Rod Groleau’s (founder of RJG Inc.) white paper, “Location Independent PPAP Streamlined for Global Manufacturing” (presented at the University of Michigan Automotive Conference in August 2000), could also apply to the medical device industry. As a collaborative team, the MOEMC exercised a Decoupled 2 (DC2) process using available systematic molding principles and technology on four predetermined, capable machines—all of different make, model (electric/hydraulic), tonnage, barrel size, volumetric injection rates, and in different locations. The concept was to challenge the traditional philosophy of a fixed machine settings validation and move toward a more flexible “Part Process Development/Validation” for a molded part across multiple machines. The MOEMC focused on reproducing a consistent “end result” based on the transfer of a DC2 process via machine independent variables (MIV) and verification of what was happening in the cavity using technology, instead of machine inputs/settings for a defined process.
The event took into consideration the applicable regulatory requirements outlined in 21 CFR part 820 as it relates to molded plastic components typically assembled into a finished device. The end result was a well-documented case study to expose the medical device industry to the “Part Process Development/Validation” strategy as a robust alternate methodology to traditional validation concepts.
The team met in May 2016 to investigate and exercise a good/better/best approach to facilitate the validation/transfer. The four mold/machine transfer trials were completed by March 2017.
Workflow
The MOEMC not only took the time to execute the mold transfer between the four distinctly different machines, it also validated the process with data-driven results and verified it using technology. The MOEMC team established a Validation Master Plan (VMP) for the event and executed the initial full validation, followed by three separate qualification/verification trials, each with its own validation report. All information was collated in a Summary Report documenting the conclusive evidence that the MIV process was repeatable and all the dimensional inspections exceeded the targeted 1.5 Ppk and attribute data (Figure 1).
Figure 1: Four validation trials were completed. Dimensional results (beginning/middle/end of each trial) were within validation control limits. Image courtesy of RJG Inc.
Pre-trial assumptions were established with a systematic process development approach that defines a “part process” paired with a “capable” machine. The focus was on optimization of what the plastic was experiencing at the part level as a result of the applied design principles, material selection, mold design and construction, and machine capability to deliver the same end results.
The 1.4g part (drive clutch) was molded in an Acetal grade material. The mold that produced the part was a 16-cavity, direct valve gate, hot runner mold with an A-side lifter and B-side actions. Once the MIV transferred process was stable for each individual trial, sample parts were measured to confirm dimensional acceptance. Technology was then utilized to visually verify and confirm that the process was normalized between the qualification and validation machines. The machine(s) were instrumented with pressure sensors and linear transducers to monitor plastic pressure and linear displacement in the injection unit. The mold had flow meters and temperature sensors to monitor the water cooling circuits and temperature of the cavity/core steel. Additionally, each cavity was equipped with force sensors to read plastic pressure. The additional data provided by technology increased confidence that the original validated process was being replicated in Trials 2 through 4.
The technology used (eDART System) allowed the team to characterize the process and graphically visualize aspects that would never otherwise be known. Documented record (objective evidence/proof) of the interaction of the outputs/results from the “plastic’s point of view” illustrated that the part process was repeatable and consistent over time, including the full history of how and why. This provided data for a visual, data-driven, quantifiable, and tangible IQ, OQ, PQ. Electronic files retain the trail of how one arrived at the process development validation and the objective evidence to correlate the part process plastic conditions to the resultant dimensional data/report (Figure 2).
Figure 2: Validation run: Process overlay of PQ, OQ High, OQ Low. Technology allowed the team to identify the upper/lower part process control limits. Image courtesy of RJG Inc.
The Results
Based on the documented results, the team has the confidence to utilize the established method as an alternative to the traditional single machine validation approach, which will be exercised and put into practice. The first article inspection report (FAIR) dimensional data for the reports for the validation and three qualification runs was executed by 3D ProScan, a division of NyproMold, using a CT scanner that compares the actual part to the 3D model and yields highly accurate results days to weeks faster than traditional metrology methods. Process capability data was measured through standard inspection practices.
The MOEMC team effectively validated and verified that the drive clutch is capable (and ready) of running production in four different machines. There is measured and documented data, representing both dimensional and process matched MIV that is supported by the data. The method and results were documented in the Validation Master Plan (VMP), and original validation report VAL-RJG-001, qualification reports VAL-RJG-002 through -004, and summary report VAL-RJG-005.
A validation (typically defined by a part dim report) has to include the molding process itself that produced the parts. This will include the record/documentation of how it was developed—proof that it is capable of producing repeatable parts (to dim) and consistently within the established control limits. Having the full roadmap, from development to production, allows a plastic part process to be portable (dimensions are a result of the defined part process). Everything is selected for a reason and should have background data to support it. That represents a true “part process” development.
Fully documented dimensional and MIV part process-related results satisfy the criteria that is typically outlined in any Master Validation Plan (MVP). The proven results are transferable to any machine that can be verified to be capable of consistently delivering the repeatable MIV plastic variables and plastic conditions. The performance of the machine has to be documented/confirmed to be within the typical recommended target limits (i.e., acceptable shot capacity, consistent melt preparation, can hold consistent part tonnage, delivers sufficient volumetric injection rate, and is not pressure limited).
These results can be plugged into any standard validation/qualification report to satisfy the regulatory documentation requirements.
The MIV checklist becomes the guidance to making a good part. Machine adjustments (from machine to machine) can be made to stay within the acceptable tolerances (established during OQ) of the defined part process. The MIV represents the plastics variables/conditions of the “part process”: a combination of the specific input and output values that characterize the particular part/mold. If these are reproduced in a defined capable machine, the same end results will be generated (i.e., part dimensions).
The use of technology provides additional process verification that the same parts are being made. It also provides a full shot-to-shot graphical record of the process outputs that represent the “fingerprint” of the part. Using a combination of the MIV Checklist and available technology (in this case, the eDART System), one can easily and quickly reproduce the process in any machine (defined as capable). This now allows for the ability to transfer molds and increases flexibility in capacity management.
Part process development/validation truly requires the engineer to look at the process from the plastic’s point of view, not the machine’s. With a machine independent approach, the machine becomes an interchangeable component of the manufacturing cell with the mold (part) at the center. The bill of materials (BOM) of the cell is comprised of the mold, machine, material, and ancillary equipment to support it. This is typically comprised of a mold temperature control unit, hot runner controller, flow meters, material dryer, robot, EOA, etc., which have all been calibration certified to performance—a typical part of the required IQ (Figure 3).
Figure 3: Compatibility: Mold and machine match based on performance specification: BOM-capable machines are interchangeable from…the plastic’s point of view. Image courtesy of RJG Inc.
Additionally, if there is a means to monitor and verify this performance, it should be done. This is required to increase confidence levels, leading to a higher level of assurance. Now during a transfer, companies can effectively run an abbreviated PQ trial and produce with confidence. Once the formal PQ report is generated and the parts pass the VMP criteria, the parts can be approved and released into the product stream.
Using a lead measure principles approach (predictive and influenceable) versus a lag approach (checking after the fact) is comprehensive and non-restrictive. This effective principle-based systematic approach to process development and validation, based on the MIV and verification with technology, can increase the level of confidence that good parts are being introduced to the product stream.
Through this new strategy, costs and resources can be significantly reduced without impacting plastic part quality or functionality. The approach requires a fully validated part process for a mold and machine(s) with detailed development history. The approach allows reduced validation during transfer to a capable machine to improve capacity restraints within manufacturing or to a new supplier.
The ultimate paradigm shift is to focus on the plastic part process outputs (plastic conditions and component variables/attributes) versus machine set points. The focus becomes the part inside the mold, not the machine, with an objective to achieve more consistent, repeatable results. Although not required, technology provides immediate verification to the originally validated part process or immediate response to normalize plastic conditions and machine performance across multiple machines. Technology can capture shot-to-shot performance, reducing reaction time associated with part measurement and visual inspection to confirm part quality (Table).
Table: A comparison of traditional mold transfer validation characteristics and
part process validation concepts.
Conclusion
Through the execution of the discovery exercise, the MOEMC has provided a broadened perspective on the acceptance of the “part process” method to allow the transfer of molds in the medical device industry as a practical alternate standard.
What was amazing to watch was that the SQE/Quality Team really dug in to learn about the molding process and its effect on the dimensional results. At the end of the Consortium, they were the most passionate about explaining the benefits of the newfound data-driven approach. Each OEM understands that they will need to execute this method (with or without technology) with one of their leading molding/converter partners to further educate internal/external teams on the principles that need to be followed.
In turn, embedding the MIV method delivers a more thorough part process development/validation that is repeatable and capable of being transferred through multiple machines. It is critical to have both the dimensional and plastic process variable (MIV) data fully documented for assurance of consistent results. Creating a case study that incorporates each OEM’s own validation protocol documentation/reporting requirements will provide a template for future program success.
The industry as a whole can now become more knowledgeable about what the levels of operational excellence (number of buildings and equipment doesn’t define this) really means when assessing a partner for manufacturing. Also, there is an increased assurance that the methods used can help reduce and mitigate risk based on data-driven decisions (using available technology) that can help drive continuous improvements and increased statistical control.
[Author’s Note: A medical device industry white paper on the subject matter is now available for download. Click here to get the white paper.]
Matt Therrien is the northeast regional manager at RJG Inc.