Matthew R Jorgensen, Ph.D., and Thor Rollins, BS, Nelson Laboratories; Allison Komiyama, Ph.D., RAC, AcKnowledge RS04.03.17
It has become more prevalent and acceptable to use the results of chemistry testing to assess the overall biocompatibility of medical devices. For many devices with prolonged or permanent patient contact, chemistry followed by toxicological assessment can provide a cost and time saving alternative to in vivo tests such as chronic, sub-chronic, genetic, and carcinogenicity testing. Manufacturers preparing to submit their device for approval by the U.S. Food and Drug Administration (FDA) rely on ISO 10993 standards and FDA guidance on the application of those standards in the planning of their chemistry testing. When it comes to the details of chemistry testing, however, the ISO standards are often vague. Furthermore, the recommendations regarding the appropriate level of testing rigor can vary within FDA. The following offers a brief overview of the framework provided by ISO 10993 guidance documents for extractable/leachable (E/L) testing, as well as three testing strategies that satisfy both ISO 10993 and FDA requirements.
There are three parts of ISO 10993 that are applicable to E/L testing:
Considering that devices commonly contain (intentionally or not) both polar and non-polar substances, and the body is composed of both polar and non-polar fluids and membranes, extraction with both polar and non-polar solvents is required. Water or saline as the polar extraction vehicle and hexane as the non-polar extraction vehicle are commonly accepted choices.
Although E/L is often used as a substitute for long-term in vivo biocompatibility tests, results are generally considered concurrently with other tests such as cytotoxicity, sensitization, and irritation. To allow comparison to these other tests, similar extraction conditions are recommended, which represent worse-case conditions per ISO 10993-12. Extraction at 50 degrees Celsius for 72 hours is a common first choice. If follow-up testing is required, other conditions more representative of clinical use can be used with justification. Extraction volume should be selected to meet several conditions: the volume should be small enough (resulting in a concentration that is high enough) so the sensitivity of the analysis in terms of µg of analyte per device is acceptable. Additionally, the extraction volume per device should be the same or less than what was used for biocompatibility testing, but still provides enough volume to be practical for all the required analytical tests.
As mentioned previously, the analysis should screen for as many compounds as possible, as it can be difficult to predict what substances could be on or in a device before analysis. To that end, the analysis should look for both organic and inorganic compounds, be able to identify these compounds, and be able to measure their concentration. Mass spectrometry methods are able to do all of these. Inductively coupled plasma mass spectrometry (ICP/MS) identifies and measures metals (which addresses inorganic compounds), while gas chromatography mass spectrometry (GC/MS) methods identify different classes of organic compounds (provided the molecules are not too large), and liquid chromatography mass spectrometry is used to screen for any organic molecules too large for GC/MS. Therefore, to cast as wide a net as possible, the extracts and their blanks should be analyzed using each of these methods. For a single device, analysis of four liquid samples is involved (one polar, one non-polar, and two blanks), creating a total of 12 mass spectrometry analyses per device. A typical testing matrix is shown in Table 1.

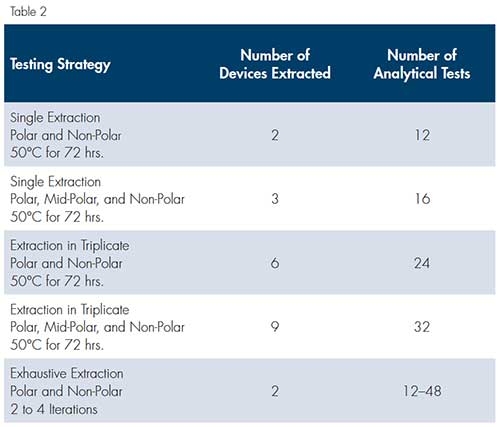
The analysis in Table 1 represents the most basic E/L analysis strategy: two solvents and two blanks, undergoing a total of 12 analytical tests. Recently, the FDA has requested variations on this analytical strategy that result in increased rigor of the analysis. For example, there have been requests for the addition of a third mid-polarity solvent, such as isopropanol, for the analysis to be repeated in triplicate, or for exhaustive extraction methods to be used. Exhaustive extraction involves repeating the basic extraction and analysis until the currently detected extracts are less than 10 percent of the sum of all extracts; this strategy involves, at a minimum, two analyses but could include more. These strategies are compared in Table 2.
The cost and turn-around time of E/L testing depends on the lab, but generally scales with the number of analytical tests requested or required. Practices and level of rigor can vary widely in labs performing E/L testing due to the ambiguity of ISO 10993-18. It is critical to determine if the level of rigor meets what is required for toxicological risk assessment per ISO 10993-17. One of the largest variables is whether detected analytes are rigorously quantitated or if they have concentrations that are estimated using semi-quantitative methods. Often, the reality is a combination of both. For example, a lab may report some analytes quantitatively if they are on a panel of common compounds, but only estimate others in a single report. For each analytical method used by a lab, the level of method validation can also vary. Questions of analytical rigor come into play when the chemistry results are interpreted by a toxicologist. If the expert interpreting the results is skilled, they may notice deficiencies in the analytical methodology and be unable to draw a conclusion or make a recommendation other than “more chemistry testing is needed.”
Selection of a testing strategy should be primarily based upon any specific feedback received from FDA. If no feedback is available, several factors should be considered: the device type, the nature and duration of patient contact, how conservative of an approach is desired, and the cost. FDA may request E/L testing above and beyond the typically required biocompatibility program. For example, we have seen requests for E/L on devices with brightly colored parts, especially when the targeted patient population includes neonates or other sensitive groups. There has also been a trend in requests for exhaustive E/L testing from FDA for devices with respiratory patient contact.
Approaching E/L chemistry testing can be a daunting task to the uninitiated; there are many variables and the guidance provided by the ISO 10993 standards and FDA is vague. However, a basic analytical protocol (shown in Table 1) has been widely accepted along with extraction conditions that mirror those familiar to biocompatibility testing. Ultimately, the most sure-fire approach is to develop a testing plan in collaboration with the toxicologist interpreting the results and FDA. The recently released FDA guidance document on the use of ISO 10993 stresses the importance of a written biocompatibility evaluation plan (BEP). In this plan, the potential risks of the device should be discussed, as well as the approaches to address the potential risks. The BEP is an excellent place to outline the strategy proposed for E/L chemical analysis and allow FDA to review and comment on the plan as part of a pre-submission conversation. With a nod of approval from FDA and the toxicologist, you can proceed confidently into the world of chemistry.
Matthew R. Jorgensen, Ph.D., is a chemistry and materials scientist with Nelson Laboratories. Thor Rollins, B.S., RN (NRCM) is a senior scientist with the company. Allison Komiyama, Ph.D., RAC, is owner and principal consultant at AcKnowledge Regulatory Strategies.
There are three parts of ISO 10993 that are applicable to E/L testing:
- 10993-12: How to prepare the sample—Defines what an extractable is versus a leachable, the criteria for exhaustive extraction, recommendations on extraction volume and extraction vehicles (polar and non-polar).
- 10993-18 (BE83:2006/(R)2011): How to analyze the sample—Provides general guidance on the types of analytical instruments appropriate for identification and quantification of different analytes.
- 10993-17: How to interpret the results—Provides guidance on how to establish allowable limits (tolerable exposures) for substances extracted from medical devices.
Considering that devices commonly contain (intentionally or not) both polar and non-polar substances, and the body is composed of both polar and non-polar fluids and membranes, extraction with both polar and non-polar solvents is required. Water or saline as the polar extraction vehicle and hexane as the non-polar extraction vehicle are commonly accepted choices.
Although E/L is often used as a substitute for long-term in vivo biocompatibility tests, results are generally considered concurrently with other tests such as cytotoxicity, sensitization, and irritation. To allow comparison to these other tests, similar extraction conditions are recommended, which represent worse-case conditions per ISO 10993-12. Extraction at 50 degrees Celsius for 72 hours is a common first choice. If follow-up testing is required, other conditions more representative of clinical use can be used with justification. Extraction volume should be selected to meet several conditions: the volume should be small enough (resulting in a concentration that is high enough) so the sensitivity of the analysis in terms of µg of analyte per device is acceptable. Additionally, the extraction volume per device should be the same or less than what was used for biocompatibility testing, but still provides enough volume to be practical for all the required analytical tests.
As mentioned previously, the analysis should screen for as many compounds as possible, as it can be difficult to predict what substances could be on or in a device before analysis. To that end, the analysis should look for both organic and inorganic compounds, be able to identify these compounds, and be able to measure their concentration. Mass spectrometry methods are able to do all of these. Inductively coupled plasma mass spectrometry (ICP/MS) identifies and measures metals (which addresses inorganic compounds), while gas chromatography mass spectrometry (GC/MS) methods identify different classes of organic compounds (provided the molecules are not too large), and liquid chromatography mass spectrometry is used to screen for any organic molecules too large for GC/MS. Therefore, to cast as wide a net as possible, the extracts and their blanks should be analyzed using each of these methods. For a single device, analysis of four liquid samples is involved (one polar, one non-polar, and two blanks), creating a total of 12 mass spectrometry analyses per device. A typical testing matrix is shown in Table 1.
The analysis in Table 1 represents the most basic E/L analysis strategy: two solvents and two blanks, undergoing a total of 12 analytical tests. Recently, the FDA has requested variations on this analytical strategy that result in increased rigor of the analysis. For example, there have been requests for the addition of a third mid-polarity solvent, such as isopropanol, for the analysis to be repeated in triplicate, or for exhaustive extraction methods to be used. Exhaustive extraction involves repeating the basic extraction and analysis until the currently detected extracts are less than 10 percent of the sum of all extracts; this strategy involves, at a minimum, two analyses but could include more. These strategies are compared in Table 2.
The cost and turn-around time of E/L testing depends on the lab, but generally scales with the number of analytical tests requested or required. Practices and level of rigor can vary widely in labs performing E/L testing due to the ambiguity of ISO 10993-18. It is critical to determine if the level of rigor meets what is required for toxicological risk assessment per ISO 10993-17. One of the largest variables is whether detected analytes are rigorously quantitated or if they have concentrations that are estimated using semi-quantitative methods. Often, the reality is a combination of both. For example, a lab may report some analytes quantitatively if they are on a panel of common compounds, but only estimate others in a single report. For each analytical method used by a lab, the level of method validation can also vary. Questions of analytical rigor come into play when the chemistry results are interpreted by a toxicologist. If the expert interpreting the results is skilled, they may notice deficiencies in the analytical methodology and be unable to draw a conclusion or make a recommendation other than “more chemistry testing is needed.”
Selection of a testing strategy should be primarily based upon any specific feedback received from FDA. If no feedback is available, several factors should be considered: the device type, the nature and duration of patient contact, how conservative of an approach is desired, and the cost. FDA may request E/L testing above and beyond the typically required biocompatibility program. For example, we have seen requests for E/L on devices with brightly colored parts, especially when the targeted patient population includes neonates or other sensitive groups. There has also been a trend in requests for exhaustive E/L testing from FDA for devices with respiratory patient contact.
Approaching E/L chemistry testing can be a daunting task to the uninitiated; there are many variables and the guidance provided by the ISO 10993 standards and FDA is vague. However, a basic analytical protocol (shown in Table 1) has been widely accepted along with extraction conditions that mirror those familiar to biocompatibility testing. Ultimately, the most sure-fire approach is to develop a testing plan in collaboration with the toxicologist interpreting the results and FDA. The recently released FDA guidance document on the use of ISO 10993 stresses the importance of a written biocompatibility evaluation plan (BEP). In this plan, the potential risks of the device should be discussed, as well as the approaches to address the potential risks. The BEP is an excellent place to outline the strategy proposed for E/L chemical analysis and allow FDA to review and comment on the plan as part of a pre-submission conversation. With a nod of approval from FDA and the toxicologist, you can proceed confidently into the world of chemistry.
Matthew R. Jorgensen, Ph.D., is a chemistry and materials scientist with Nelson Laboratories. Thor Rollins, B.S., RN (NRCM) is a senior scientist with the company. Allison Komiyama, Ph.D., RAC, is owner and principal consultant at AcKnowledge Regulatory Strategies.