Medtronic07.31.18
Medtronic plc has received U.S. Food and Drug Administration (FDA) approval for the Implantable System for Remodulin (ISR) to treat patients with pulmonary arterial hypertension (PAH). Through a first-of-its-kind collaboration, the Medtronic SynchroMed II drug delivery system and cardiac catheter technologies were leveraged to deliver the PAH medication Remodulin (treprostinil) Injection developed by United Therapeutics Corporation. United Therapeutics will lead the commercial promotion of the ISR, with Medtronic support.
PAH is a severely debilitating and progressive disease that causes high blood pressure in the pulmonary arteries, ultimately resulting in right-heart failure and premature death. It predominantly affects women, who are typically diagnosed in their late 30s to early 50s.1,2,3
"External infusion pumps have been used to deliver prostacyclins for PAH, but managing the therapy places a significant burden on patients, interferes with their daily activities, and runs a high risk of infections," said David Steinhaus, M.D., general manager of the Heart Failure business, part of the Cardiac and Vascular Group at Medtronic. "This fully implantable drug delivery system was designed to address these serious patient care concerns."
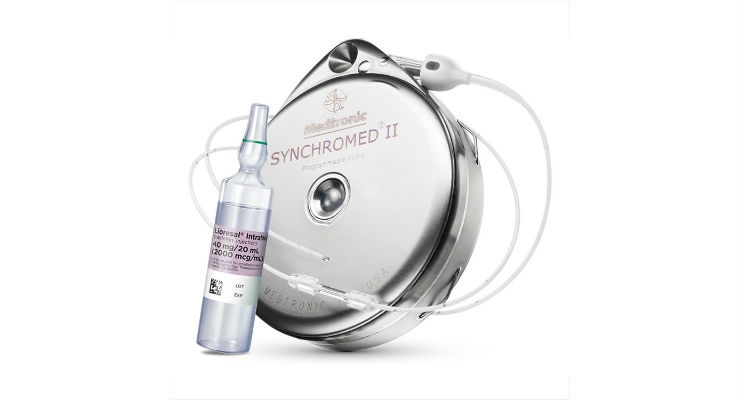
The system is composed of the Company's SynchroMed II implantable drug infusion pump and a newly developed intravascular catheter to deliver Remodulin intravenously to patients who have previously been receiving Remodulin intravenously via an external infusion pump. Medtronic and United Therapeutics pursued parallel regulatory filings for the device and drug, respectively.
FDA approval was based on the DelIVery for PAH trial, a prospective, single-arm, non-randomized, open-label study conducted at 10 sites in the United States. It enrolled 64 patients (60 successfully implanted) and showed the implantable intravascular delivery system effectively delivered treprostinil, with a low rate of catheter-related complications, and a high rate of patient satisfaction.4
In collaboration with leading clinicians, researchers and scientists worldwide, Medtronic offers the broadest range of innovative medical technology for the interventional and surgical treatment of cardiovascular disease and cardiac arrhythmias. The company strives to offer products and services of the highest quality that deliver clinical and economic value to healthcare consumers and providers around the world.
References
1 Thenappan T, Shah SJ, Rich S, Gomberg-Maitland M. A USA-based registry for pulmonary arterial hypertension: 1982-2006. Eur Respir J. 2007;30:1103-10
2 Humbert M. Update in pulmonary arterial hypertension. 2007. Am J Respir Crit Care Med. 2008;177:574-9
3 Runo JR, Loyd JE. Primary pulmonary hypertension. Lancet. 2003;361(9368):1533-44.
4 Bourge RC, Waxman AB, Gomberg-Maitland M, et al. Treprostinil administered to treat pulmonary arterial hypertension using a fully implantable programmable intravascular delivery system: Results of the Delivery for PAH trial. Chest. 2016;150(1):27-34.
PAH is a severely debilitating and progressive disease that causes high blood pressure in the pulmonary arteries, ultimately resulting in right-heart failure and premature death. It predominantly affects women, who are typically diagnosed in their late 30s to early 50s.1,2,3
"External infusion pumps have been used to deliver prostacyclins for PAH, but managing the therapy places a significant burden on patients, interferes with their daily activities, and runs a high risk of infections," said David Steinhaus, M.D., general manager of the Heart Failure business, part of the Cardiac and Vascular Group at Medtronic. "This fully implantable drug delivery system was designed to address these serious patient care concerns."
The system is composed of the Company's SynchroMed II implantable drug infusion pump and a newly developed intravascular catheter to deliver Remodulin intravenously to patients who have previously been receiving Remodulin intravenously via an external infusion pump. Medtronic and United Therapeutics pursued parallel regulatory filings for the device and drug, respectively.
FDA approval was based on the DelIVery for PAH trial, a prospective, single-arm, non-randomized, open-label study conducted at 10 sites in the United States. It enrolled 64 patients (60 successfully implanted) and showed the implantable intravascular delivery system effectively delivered treprostinil, with a low rate of catheter-related complications, and a high rate of patient satisfaction.4
In collaboration with leading clinicians, researchers and scientists worldwide, Medtronic offers the broadest range of innovative medical technology for the interventional and surgical treatment of cardiovascular disease and cardiac arrhythmias. The company strives to offer products and services of the highest quality that deliver clinical and economic value to healthcare consumers and providers around the world.
References
1 Thenappan T, Shah SJ, Rich S, Gomberg-Maitland M. A USA-based registry for pulmonary arterial hypertension: 1982-2006. Eur Respir J. 2007;30:1103-10
2 Humbert M. Update in pulmonary arterial hypertension. 2007. Am J Respir Crit Care Med. 2008;177:574-9
3 Runo JR, Loyd JE. Primary pulmonary hypertension. Lancet. 2003;361(9368):1533-44.
4 Bourge RC, Waxman AB, Gomberg-Maitland M, et al. Treprostinil administered to treat pulmonary arterial hypertension using a fully implantable programmable intravascular delivery system: Results of the Delivery for PAH trial. Chest. 2016;150(1):27-34.