James A. Dunning, Owner, QPC Services LLC10.05.17
The medical device and pharmaceutical industries are both very unique markets. Although they share the same goal—improving health—the two are different in obvious ways: mechanical (device) vs. chemical (drugs), internal vs. external, volume (over 500,000 types of medical devices compared with 20,000 medicines), and product development timelines.
There is, of course, common ground between the industries—namely, hefty profit margins, pricing pressures, product promotion, and perhaps most importantly, the U.S. Food and Drug Administration. Regulations may not seem like an area of mutual harmony between the two, but I contend otherwise. I believe there are more similarities between the two industries than meets the eye; hence my motivation for this ongoing series comparing parts of the drug and device industries’ Current Good Manufacturing Practices (cGMP) requirements.
This month’s column will focus on ways 21 CFR Part 820 Quality System Regulation, also known as the cGMP regulation for medical devices, aligns with the cGMP requirements of 21 CFR Part 211 Subpart C—Buildings and Facilities, and Subpart D—Equipment. Last month’s article dealt with Subpart B—Organization and Personnel. As was the case in previous article, the term “pharmaceutical(s)” is used rather than “drug(s)” unless the term is part of an excerpt from the cGMP regulation. Scope and definition are the same as well.
An overview of the scope of this article is provided in Table 1.
Table 1 shows Pharmaceutical cGMP requirements for Buildings and Facilities are more specific than those for medical devices—the Pharmaceutical cGMP, for example, has a clause specifically for Lighting. Yet the Medical Device cGMP mandates are no less comprehensive. Unlike its counterpart, which demands adequate lighting be provided “in all areas,” the Medical Device cGMP requirements indirectly address the subject through 820.70(c) Environmental control: “Where environmental conditions could reasonably be expected to have an adverse effect on product quality, the manufacturer shall establish and maintain procedures to adequately control these environmental conditions. Environmental control system(s) shall be periodically inspected to verify that the system, including necessary equipment, is adequate and functioning properly. These activities shall be documented and review.” Thus, it is reasonable to conclude both cGMPs include an adequate lighting mandate.
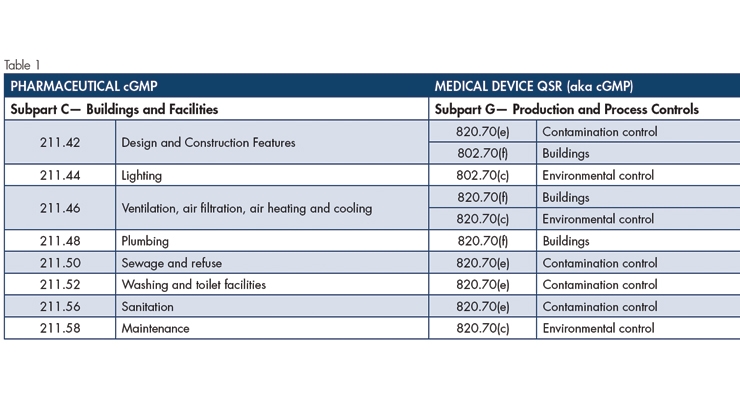
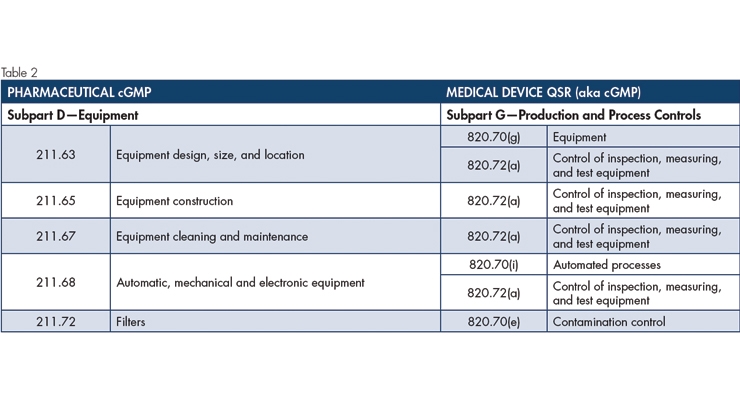
Table 2 shows the Pharmaceutical cGMP is more specific for equipment. But as with lighting, the device cGMP equipment requirements should not be discounted due to wording (or lack thereof). The drug industry’s mandate for filters (under equipment) states, “Filters for liquid filtration used in the manufacture, processing, or packing of injectable drug products intended for human use shall not release fibers into such products. Fiber-releasing filters may be used when it is not possible to manufacture such products without the use of these filters. If use of a fiber-releasing filter is necessary, an additional nonfiber-releasing filter having a maximum nominal pore size rating of 0.2 micron (0.45 micron if the manufacturing conditions so dictate) shall subsequently be used to reduce the content of particles in the injectable drug product. The use of asbestos-containing filter is prohibited.” Asbestos and fiber-releasing filters are not specifically addressed in the Medical Device cGMP, but the contamination control requirement reads, “Each manufacturer shall establish and maintain procedures to prevent contamination of equipment or product by substances that could reasonably be expected to have an adverse effect on product quality.” Although it does not mention fibers or asbestos, it is reasonable to conclude the device cGMP mandates would not allow for fiber-releasing filters to make product contact.
In conclusion, I believe it is fair to say both industries’ cGMP requirements are equivalent regarding Buildings and Facilities, and Equipment.
Further Reading
Review any of the other sections of this series by clicking on the headline.
Current Good Manufacturing Practices: Pharma vs. Device
Comparing cGMP Pharma vs. Device: Subpart A – General Provisions
Comparing cGMP Pharma vs. Device: Subpart A—General Provisions (Part II)
Comparing cGMP Pharma vs. Device: Subpart B—Organization and Personnel
Comparing cGMP Pharma vs. Device: Subpart E—Control of Components, Containers and Closures
James A. “Jim” Dunning’s consulting career began in 2001. He has provided quality and regulatory consulting services for various companies ranging from Fortune 500 medical device firms to startups. Dunning’s passion, however, lies with startups and small companies, especially those in regulatory distress. He is experienced in preparing 510(k) applications, developing complete Quality Management Systems, providing Quality System Training, and advising on quality, business, and leadership issues. Dunning is a senior member of the American Society for Quality (ASQ) and a member of the Regulatory Affairs Professional Society (RAPS). He can be reached at jdunning@qpcservices.com
There is, of course, common ground between the industries—namely, hefty profit margins, pricing pressures, product promotion, and perhaps most importantly, the U.S. Food and Drug Administration. Regulations may not seem like an area of mutual harmony between the two, but I contend otherwise. I believe there are more similarities between the two industries than meets the eye; hence my motivation for this ongoing series comparing parts of the drug and device industries’ Current Good Manufacturing Practices (cGMP) requirements.
This month’s column will focus on ways 21 CFR Part 820 Quality System Regulation, also known as the cGMP regulation for medical devices, aligns with the cGMP requirements of 21 CFR Part 211 Subpart C—Buildings and Facilities, and Subpart D—Equipment. Last month’s article dealt with Subpart B—Organization and Personnel. As was the case in previous article, the term “pharmaceutical(s)” is used rather than “drug(s)” unless the term is part of an excerpt from the cGMP regulation. Scope and definition are the same as well.
An overview of the scope of this article is provided in Table 1.
Table 1 shows Pharmaceutical cGMP requirements for Buildings and Facilities are more specific than those for medical devices—the Pharmaceutical cGMP, for example, has a clause specifically for Lighting. Yet the Medical Device cGMP mandates are no less comprehensive. Unlike its counterpart, which demands adequate lighting be provided “in all areas,” the Medical Device cGMP requirements indirectly address the subject through 820.70(c) Environmental control: “Where environmental conditions could reasonably be expected to have an adverse effect on product quality, the manufacturer shall establish and maintain procedures to adequately control these environmental conditions. Environmental control system(s) shall be periodically inspected to verify that the system, including necessary equipment, is adequate and functioning properly. These activities shall be documented and review.” Thus, it is reasonable to conclude both cGMPs include an adequate lighting mandate.
Table 2 shows the Pharmaceutical cGMP is more specific for equipment. But as with lighting, the device cGMP equipment requirements should not be discounted due to wording (or lack thereof). The drug industry’s mandate for filters (under equipment) states, “Filters for liquid filtration used in the manufacture, processing, or packing of injectable drug products intended for human use shall not release fibers into such products. Fiber-releasing filters may be used when it is not possible to manufacture such products without the use of these filters. If use of a fiber-releasing filter is necessary, an additional nonfiber-releasing filter having a maximum nominal pore size rating of 0.2 micron (0.45 micron if the manufacturing conditions so dictate) shall subsequently be used to reduce the content of particles in the injectable drug product. The use of asbestos-containing filter is prohibited.” Asbestos and fiber-releasing filters are not specifically addressed in the Medical Device cGMP, but the contamination control requirement reads, “Each manufacturer shall establish and maintain procedures to prevent contamination of equipment or product by substances that could reasonably be expected to have an adverse effect on product quality.” Although it does not mention fibers or asbestos, it is reasonable to conclude the device cGMP mandates would not allow for fiber-releasing filters to make product contact.
In conclusion, I believe it is fair to say both industries’ cGMP requirements are equivalent regarding Buildings and Facilities, and Equipment.
Further Reading
Review any of the other sections of this series by clicking on the headline.
Current Good Manufacturing Practices: Pharma vs. Device
Comparing cGMP Pharma vs. Device: Subpart A – General Provisions
Comparing cGMP Pharma vs. Device: Subpart A—General Provisions (Part II)
Comparing cGMP Pharma vs. Device: Subpart B—Organization and Personnel
Comparing cGMP Pharma vs. Device: Subpart E—Control of Components, Containers and Closures
James A. “Jim” Dunning’s consulting career began in 2001. He has provided quality and regulatory consulting services for various companies ranging from Fortune 500 medical device firms to startups. Dunning’s passion, however, lies with startups and small companies, especially those in regulatory distress. He is experienced in preparing 510(k) applications, developing complete Quality Management Systems, providing Quality System Training, and advising on quality, business, and leadership issues. Dunning is a senior member of the American Society for Quality (ASQ) and a member of the Regulatory Affairs Professional Society (RAPS). He can be reached at jdunning@qpcservices.com