
Business Wire06.05.18
Apollo Endosurgery Inc. a developer of less invasive medical devices for bariatric and gastrointestinal procedures, announced that the United States Food and Drug Administration (FDA) has approved updates to the ORBERA Intragastric Balloon System’s U.S. labeling including a new Physician Directions for Use (DFU), Physician Training, and Patient Directions for Use. Apollo, working directly with the Food and Drug Administration, has developed these updates as a means of communicating ORBERA’s most current safety information to both physicians and patients. The Letter to Healthcare Providers issued by the FDA is intended to highlight the updated labeling. The new DFU is available to physicians and patients here.
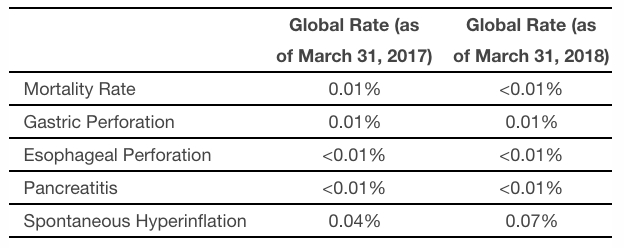
Apollo’s last ORBERA labeling update occurred on June 6, 2017 and included information regarding adverse events of acute pancreatitis and spontaneous hyperinflation, which were not seen during the ORBERA United States pivotal study. The newly revised and approved labeling announced today provides updates for these two events and also adds information regarding the risks of gastric and esophageal perforation, aspiration, and death. The table below summarizes the global rate of occurrence for these adverse events as included in the revised DFU (data as of March 31, 2018) and in the previous DFU (data as of March 31, 2017).
Also included in this DFU update are specific U.S. rates for acute pancreatitis, spontaneous hyperinflation, gastric and esophageal perforation, and aspiration. The newly approved DFU also separately reports a U.S. mortality rate in ORBERA patients of 0.036% (i.e. less than 4 deaths per 10,000 patients) to provide additional awareness to U.S. physicians and patients.
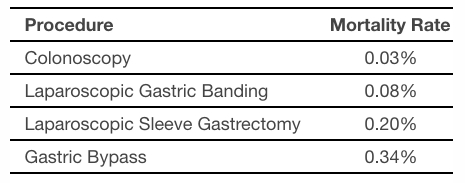
The table below provides mortality rates for procedures performed in gastrointestinal and bariatric medicine as reported in peer-review literature. The mortality rate of a colonoscopy, which is the most common procedure performed in gastrointestinal medicine, is in line with that of ORBERA. It is also important to understand that colonoscopies are generally performed as a screening procedure, while ORBERA is an actual treatment procedure.
The U.S. Physician Training has also been revised and approved by the FDA. The updated training provides additional guidance on management of ORBERA patients experiencing symptoms suggestive of persistent intolerance (identified as “refractory intolerance” in the revised labeling). While infrequent in occurrence, these patients can develop an intolerance to the balloon potentially caused by a greater than anticipated delay in gastric emptying with gastric distention and retention of food and fluid. These patients may be at greater risk for both perforation and aspiration at the time of balloon removal.
To help physicians recognize the signs and symptoms of persistent intolerance, and appropriately manage these patients, updated training has been developed to provide:
These updates will be incorporated into the patient labeling, physician labeling and physician training. All previously trained physicians will also receive this new training.
”These updates provide important enhancements to our existing labeling and support our continued emphasis on patient safety. Physicians should always monitor patients closely during the entire term of treatment, and patients should be thoroughly instructed on signs or symptoms of potentially life-threatening adverse events,” said Christopher Gostout, M.D., chief medical officer at Apollo Endosurgery.
World-wide, four (4) occurrences of death in patients who received the ORBERA Intragastric Balloon have been reported since the FDA’s “Updated Letter to Healthcare Providers” dated August 10, 2017. One (1) of these four (4) reported events occurred in the U.S. since the FDA’s August 2017 update. In analyzing the information available for these four (4) reports, gastric perforation preceded three (3) of the deaths including the U.S. event reported on 1/12/18. The links below provides summary information regarding these reports as well as access to the full MedWatch reports via a direct link to the FDA’s MAUDE database:
In each case, Apollo provided the event report to FDA through the MedWatch system and, per Apollo’s standard procedures, exhausted all efforts to obtain information directly from the treating physicians. In the absence of information that clearly states the ORBERA did not cause or contribute to the adverse event, Apollo’s standard procedure is to report the events through MedWatch.
Apollo’s last ORBERA labeling update occurred on June 6, 2017 and included information regarding adverse events of acute pancreatitis and spontaneous hyperinflation, which were not seen during the ORBERA United States pivotal study. The newly revised and approved labeling announced today provides updates for these two events and also adds information regarding the risks of gastric and esophageal perforation, aspiration, and death. The table below summarizes the global rate of occurrence for these adverse events as included in the revised DFU (data as of March 31, 2018) and in the previous DFU (data as of March 31, 2017).
Also included in this DFU update are specific U.S. rates for acute pancreatitis, spontaneous hyperinflation, gastric and esophageal perforation, and aspiration. The newly approved DFU also separately reports a U.S. mortality rate in ORBERA patients of 0.036% (i.e. less than 4 deaths per 10,000 patients) to provide additional awareness to U.S. physicians and patients.
The table below provides mortality rates for procedures performed in gastrointestinal and bariatric medicine as reported in peer-review literature. The mortality rate of a colonoscopy, which is the most common procedure performed in gastrointestinal medicine, is in line with that of ORBERA. It is also important to understand that colonoscopies are generally performed as a screening procedure, while ORBERA is an actual treatment procedure.
The U.S. Physician Training has also been revised and approved by the FDA. The updated training provides additional guidance on management of ORBERA patients experiencing symptoms suggestive of persistent intolerance (identified as “refractory intolerance” in the revised labeling). While infrequent in occurrence, these patients can develop an intolerance to the balloon potentially caused by a greater than anticipated delay in gastric emptying with gastric distention and retention of food and fluid. These patients may be at greater risk for both perforation and aspiration at the time of balloon removal.
To help physicians recognize the signs and symptoms of persistent intolerance, and appropriately manage these patients, updated training has been developed to provide:
- more detailed descriptions of the symptomology of persistent intolerance,
- methods of assessing these patients and
- updated recommendations for the management of symptoms and removal of the device.
These updates will be incorporated into the patient labeling, physician labeling and physician training. All previously trained physicians will also receive this new training.
”These updates provide important enhancements to our existing labeling and support our continued emphasis on patient safety. Physicians should always monitor patients closely during the entire term of treatment, and patients should be thoroughly instructed on signs or symptoms of potentially life-threatening adverse events,” said Christopher Gostout, M.D., chief medical officer at Apollo Endosurgery.
World-wide, four (4) occurrences of death in patients who received the ORBERA Intragastric Balloon have been reported since the FDA’s “Updated Letter to Healthcare Providers” dated August 10, 2017. One (1) of these four (4) reported events occurred in the U.S. since the FDA’s August 2017 update. In analyzing the information available for these four (4) reports, gastric perforation preceded three (3) of the deaths including the U.S. event reported on 1/12/18. The links below provides summary information regarding these reports as well as access to the full MedWatch reports via a direct link to the FDA’s MAUDE database:
- United States (Event Date: 4/4/16 — Reported Date: 4/2/18)
- Great Britain (Event Date: 11/7/16 — Reported Date: 1/3/18)
- Brazil (Event Date: 9/10/17 — Reported Date: 11/29/17)
- United States (Event Date: 1/12/18 — Reported Date: 2/12/18)
In each case, Apollo provided the event report to FDA through the MedWatch system and, per Apollo’s standard procedures, exhausted all efforts to obtain information directly from the treating physicians. In the absence of information that clearly states the ORBERA did not cause or contribute to the adverse event, Apollo’s standard procedure is to report the events through MedWatch.
