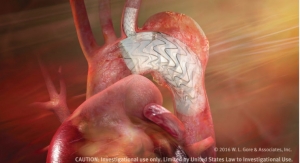
W. L. Gore & Associates Inc. has received U.S. Food and Drug Administration (FDA) approval for its Viabahn Endoprosthesis for interventional treatment of in-stent restenosis in the superficial femoral artery (SFA). Now indicated for the treatment of failed bare metal stents (BMS) in the SFA, the Viabahn device has more vascular indications supported by more Level I clinical trials than any other self-expanding peripheral stent or stent-graft, according to company officials.
In-stent restenosis—re-clogging of an artery—or reocclusion occurs in a significant number of patients over the course of one year after bare metal stenting, and rates are highly dependent on the severity of disease. Treatment options for in-stent restenosis include angioplasty, repeat stenting with another BMS, and surgical bypass. The Viabahn device changes the current treatment paradigm for in-stent restenosis by re-lining the failed BMS and adjacent diseased vessel, providing a long-term physical barrier that extends the life of the intervention, according to the company.
The safety and efficacy the device was supported by the randomized RELINE clinical study, in which the lesions in both arms of the study averaged more than 17 centimeters (cm) in length. Gore officials claim the study found that the device is statistically superior in target lesion primary patency than percutaneous transluminal angioplasty, or PTA, which is the most common frontline interventional treatment of in-stent restenosis.
The RELINE study found that subjects in the Viabahn arm of the RELINE trial were approximately three times less likely than those in the PTA arm to require a target lesion revascularization (TLR) after one year; after 12 months, Gore's device showed a primary patency of 74.8 percent, more than 45 percent higher than PTA; and with the longest available 25 cm stent-graft, heparin surface, and ePTFE (a proprietary polytetrafluoroethylene polymer made by Gore) liner, Viabahn is a long-lasting solution for treatment of failed bare stents.
“The treatment of failed bare metal stents has long been a perplexing problem for vascular specialists, in which only a few treatments are FDA-approved and limited compelling clinical data exist. This new indication for the Gore device and the accompanying data from the RELINE trial provide physicians with the means to intervene confidently on a failed bare metal stent, extending the life of the intervention and improving patient outcomes,” said Peter A. Soukas, M.D., assistant professor of medicine, Brown University Medical School in Providence, R.I.
Gore officials say it is the only stent-graft to receive approval for the SFA (de novo, restenotic, and in-stent restenotic disease), iliac artery, and arteriovenous access revision. The device is constructed with a durable, reinforced, biocompatible, expanded ePTFE liner and attached to an external nitinol stent structure. The ePTFE luminal surface of the device features the CBAS Heparin surface intended to provide sustained thromboresistance--which means resistance to blood clots.
“The Gore Viabahn Endoprosthesis remains one of the most studied stents or stent-grafts for use in the SFA,” said Ray Swinney, business unit leader for the Gore Peripheral Interventional Business. “With the successful completion of the Gore RELINE clinical study, physicians can now use this trusted device to solve the toughest challenges in the SFA and expand the long-term treatment options available for patients experiencing in-stent restenosis. FDA approval for this critical indication supports our dedication to pursuing innovative clinical solutions and our strong commitment to the physicians and patients we serve.”
Arizona-based Gore is a privately held manufacturer of vascular grafts; endovascular and interventional devices; surgical meshes for hernia and soft tissue reconstruction; staple line reinforcement materials; and sutures for use in vascular, cardiac and general surgery.